25.7: Застосування вольтамметрії
- Page ID
- 27318

Вольтамметрія знаходить застосування як для кількісного аналізу, так і для аналізу характеристик. Приклади кожного з них виділені в цьому розділі.
Кількісні програми
Вольтамметрія була використана для кількісного аналізу найрізноманітніших зразків, включаючи екологічні зразки, клінічні зразки, фармацевтичні склади, сталі, бензин та нафту.
Вибір вольтамметричної техніки
Вибір того, яку вольтамметричну техніку використовувати, залежить від характеристик зразка, включаючи очікувану концентрацію аналіта та місце розташування зразка. Наприклад, амперометрія ідеально підходить для виявлення аналітів в проточних системах, включаючи аналіз крові пацієнта in vivo або як селективний датчик для швидкого аналізу одного аналіту. Портативність амперометричних датчиків, схожих на потенціометричні датчики, також робить їх ідеальними для польових досліджень. Хоча циклічна вольтамметрія використовується для визначення концентрації аналіта, інші методи, описані в цьому розділі, краще підходять для кількісної роботи.
Імпульсна полярографія та стріппінг-вольтамметрія часто взаємозамінні. Вибір того, яку техніку використовувати, часто залежить від концентрації аналіта та бажаної точності та точності. Межі виявлення для нормальної імпульсної полярографії зазвичай знаходяться на порядку від 10 —6 М до 10 —7 М, а для диференціальної імпульсної полярографії, сходової та квадратної хвильової полярографії знаходяться між 10 —7 М та 10 —9 М, оскільки ми концентруємо аналіт у зачистки вольтамметрії, межа виявлення для багатьох аналітів становить від 10 —10 М до 10 —12 М. З іншого боку, струм при зачистки вольтамметрії набагато чутливіший, ніж імпульсна полярографія, до змін експериментальних умов, що може призвести до погіршення точності та точності . Ми також можемо використовувати імпульсну полярографію для аналізу більш широкого спектру неорганічних та органічних аналітів, оскільки немає необхідності спочатку депонувати аналіт на поверхні електрода.
Зачистка вольтамметрії також страждає від випадкових перешкод, коли два метали, такі як Cu і Zn, об'єднуються, утворюючи інтерметалідну сполуку в ртутній амальгамі. Потенціал осадження Zn. досить негативний, що будь-який Cu 2 + у зразку також відкладається в краплю ртуті або плівку, що призводить до утворення інтерметалідних сполук, таких як CuZn і CuZn 2. Під час стадії зачистки цинк в інтерметалідних сполуках смуги з потенціалами поблизу потенціалів міді, зменшуючи струм для цинку при його звичайному потенціалі та збільшуючи видимий струм для міді. Подолати цю проблему можна шляхом додавання елемента, який утворює більш міцну інтерметалідну сполуку з заважаючим металом. Таким чином, додавання Ga 3+ мінімізує інтерференцію Cu при аналізі на Zn шляхом формування інтерметалідної сполуки Cu і Ga.
Виправлення на залишковий струм
При будь-якому кількісному аналізі ми повинні коригувати сигнал аналіта для сигналів, що виникають з інших джерел. Сумарний струм, тобто, в вольтамметрії складається з двох частин: струму від окислення або відновлення аналіта, i А, і фону або залишкового струму, i r.
\[i_{t o t}=i_{A}+i_{r} \label{app1} \]
Залишковий струм, в свою чергу, має два джерела. Одним з джерел є фарадаїчний струм від окислення або зменшення слідових перешкод у зразку, i int. Іншим джерелом є зарядний струм, i ch, що супроводжує зміну потенціалу робочого електрода.
\[i_{r}=i_{\mathrm{int}}+i_{c h} \label{app2} \]
Ми можемо мінімізувати фарадаїчний струм через домішки, ретельно підготувавши зразок. Наприклад, одна важлива домішка розчиняється О 2, яка піддається двоступеневому зниженню: спочатку до Н 2 О 2 при потенціалі —0,1 В проти СКВ, а потім до Н 2 О при потенціалі —0,9 В проти СКВ. Видалення розчиненого О 2 шляхом барботування інертного газу типу N 2 через зразок виключає цю перешкоду. Після зняття розчиненого О 2 витримка ковдри N 2 над верхньою частиною розчину перешкоджає повторному надходженню O 2 в розчин.
Існує два методи компенсації залишкового струму. Одним із методів є вимірювання загального струму на потенціалах, де фарадаїчний струм аналіта дорівнює нулю, і екстраполювати його на інші потенціали. Це спосіб, показаний на малюнку 25.3.7. Однією з переваг екстраполяції є те, що нам не потрібно купувати додаткові дані. Важливим недоліком є те, що екстраполяція передбачає, що будь-яка зміна залишкового струму з потенціалом передбачувана, чого може бути і не так. Другий, і більш суворий підхід, полягає в отриманні вольтаммограми для відповідної заготовки. Залишковий струм заготовки потім віднімається із загального струму зразка.
Аналіз окремих компонентів
Аналіз зразка з одним аналітом простий, використовуючи будь-який із методів стандартизації, розглянутих у розділі 1.
Концентрацію As (III) у воді визначають за допомогою диференціальної імпульсної полярографії в 1 М HCl. Початковий потенціал встановлюється на —0.1 В проти SCE і сканується в бік більшої кількості негативних потенціалів зі швидкістю 5 мВ/с. Зниження As (III) до As (0) відбувається при потенціалі приблизно -0.44 В проти SCE. Пікові струми для набору стандартних рішень, виправлених на залишковий струм, наведені в наступній таблиці.
| [Як (III)] (мкМ) | i р (мкМ) |
|---|---|
| 1.00 | 0,298 |
| 3.00 | 0,947 |
| 6.00 | 1.83 |
| 9.00 | 2.72 |
Яка концентрація As (III) в пробі води, якщо її піковий струм дорівнює 1,37 мкА?
Рішення
Лінійна регресія дає калібрувальну криву, показану на малюнку\(\PageIndex{1}\), з рівнянням
\[i_{p}=0.0176+3.01 \times[\mathrm{As}(\mathrm{III})] \nonumber \]
Підстановка пікового струму зразка в рівняння регресії дає концентрацію As (III) як 4,49 мкм.

Багатокомпонентний аналіз
Вольтамметрія є особливо привабливою методикою для аналізу зразків, які містять два або більше аналітів. За умови, що аналіти поводяться самостійно, вольтаммограма багатокомпонентної суміші являє собою підсумовування окремих вольтаммограм кожного аналіта. Як показано на малюнку\(\PageIndex{2}\), якщо розділення між напівхвильовими потенціалами або між піковими потенціалами є достатнім, ми можемо визначити наявність кожного аналіту так, ніби він єдиний аналіт у зразку. Мінімальний поділ між напівхвильовими потенціалами або піковими потенціалами для двох аналітів залежить від декількох факторів, включаючи тип електрода і сигнал потенціалу збудження. Для нормальної полярографії поділ становить не менше ± 0,2-0,3 В, а диференціально-імпульсна вольтамметрія вимагає мінімального поділу ±0,04—0,05 В.
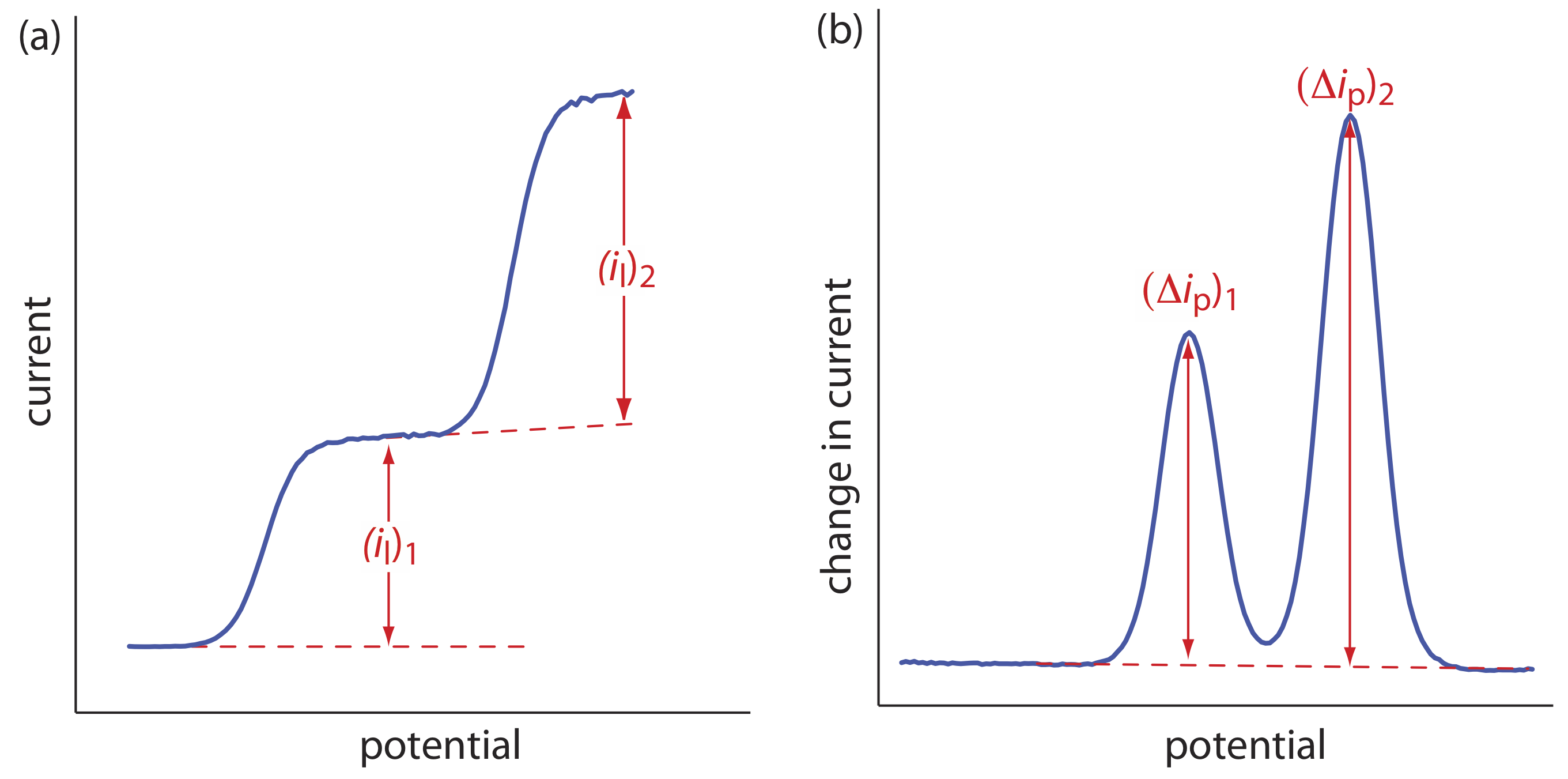
Якщо вольтаммограми для двох аналітів недостатньо розділені, можливий одночасний аналіз. Приклад такого підходу викладено на наступному прикладі.
Диференціально-імпульсний полярографічний аналіз суміші індію та кадмію в 0,1 М HCl ускладнюється перекриттям відповідних вольтаммограм [Lanza P. J. chem. Едук. 1990, 67, 704—705]. Піковий потенціал для індію - 0,557 В, а для кадмію - 0,597 В. При аналізі стандарту індію 0,800-ppm\(\Delta i_p\) (в довільних одиницях) 200,5 при —0,557 В і 87,5 при —0,597 В відносно насиченого опорного електрода Ag/AgCl. Стандартний розчин 0,793 проміле кадмію має\(\Delta i_p\) 58,5 при —0,557 В і 128,5 при —0,597 В. Яка концентрація індію і кадмію в зразку, якщо\(\Delta i_p\) становить 167,0 при потенціалі —0,557 В і 99,5 при потенціалі —0,597В.
Рішення
Зміна струму\(\Delta i_p\), в диференціальній імпульсній полярографії є лінійною функцією концентрації аналіта
\[\Delta i_{p}=k_{A} C_{A} \nonumber \]
де k A - константа, яка залежить від аналіту і прикладного потенціалу, а C A - концентрація аналіта. Щоб визначити концентрації індію та кадмію у зразку, ми повинні спочатку знайти значення k A для кожного аналіту при кожному потенціалі. Для простоти виділимо потенціал —0,557 В як Е 1, а для —0,597 В як Е 2. Значення k A є
\[\begin{aligned} k_{\mathrm{In}, E_{1}} &=\frac{200.5}{0.800 \ \mathrm{ppm}}=250.6 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{In}, E_{2}} &=\frac{87.5}{0.800 \ \mathrm{ppm}}=109.4 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{Cd} E_{1}} &=\frac{58.5}{0.793 \ \mathrm{ppm}}=73.8 \ \mathrm{ppm}^{-1} \\ k_{\mathrm{Cd} E_{2}} &=\frac{128.5}{0.793 \ \mathrm{ppm}}=162.0 \ \mathrm{ppm}^{-1} \end{aligned} \nonumber \]
Далі пишемо одночасні рівняння для струму на двох потенціалах.
\[\begin{array}{l}{\Delta i_{E_{1}}=167.0=250.6 \ \mathrm{ppm}^{-1} \times C_{\mathrm{In}}+73.8 \ \mathrm{ppm}^{-1} \times C_{\mathrm{Cd}}} \\ {\triangle i_{E_{2}}=99.5=109.4 \ \mathrm{ppm}^{-1} \times C_{\mathrm{In}}+162.0 \ \mathrm{ppm}^{-1} \times C_{\mathrm{Cd}}}\end{array} \nonumber \]
Розв'язування одночасних рівнянь, яке залишають як вправу, дає концентрацію індію як 0,606 проміле, а концентрацію кадмію - 0,205 проміле.
Екологічні зразки
Вольтамметрія є одним з декількох важливих аналітичних методів аналізу мікрометалів у зразках навколишнього середовища, включаючи підземні води, озера, річки та струмки, морську воду, дощ та сніг. Межі виявлення на рівні частин на мільярд є звичайними для багатьох слідових металів, використовуючи диференціальну імпульсну полярографію, з анодною зачисткою вольтамметрії, що забезпечує межі виявлення деталей на трильйон для деяких слідових металів.
Одним з цікавих екологічних застосувань анодної зачистки вольтамметрії є визначення хімічної форми мікрометалу в пробі води. Видоутворення має важливе значення, оскільки біодоступність, токсичність та легкість транспортування через навколишнє середовище часто залежить від його хімічної форми. Наприклад, мікрометал, який сильно пов'язаний з колоїдними частинками, як правило, не токсичний, оскільки він недоступний водним формам життя. На жаль, анодна зачистка вольтамметрії не може розрізнити точну хімічну форму сліду металу, оскільки тісно пов'язані види, такі як Pb 2 + і PbCl +, виробляють єдиний пік зачистки. Натомість слідові метали поділяються на «операційно визначені» категорії, які мають екологічне значення.
Оперативно визначена означає, що аналіт поділяється на категорії за конкретними методами, що використовуються для виділення його із зразка. У екологічній літературі багато прикладів оперативних визначень. Наприклад, розподіл слідів металів у ґрунтах та відкладеннях часто визначається з точки зору реагентів, що використовуються для їх вилучення; таким чином, ви можете знайти оперативне визначення для Zn 2 + в озерному осаду як те, що видобувається за допомогою 1,0 М ацетату натрію, або що видобувається за допомогою 1,0 М HCl.
Хоча існує багато схем видоутворення в екологічній літературі, ми розглянемо одну, запропоновану Бетлі та Флоренцією [див. (a) Батлі, Г. Е.; Флоренція, Т. М. Летт. 1976, 9, 379—388; (б) Бетлі, Г.Е.; Флоренція, Т. М. Таланта 1977, 24, 151—158; (в) Бетлі, Г.Е.; Флоренс, Т. Хім. 1980, 52, 1962—1963; (d) Флоренс, Т.М., Батлі, Г.Е.; CRC Crit. Преподобний Анальний. Хім. 1980, 9, 219—296]. Ця схема, яка викладена в табл.\(\PageIndex{2}\), поєднує анодну зачистку вольтамметрії з іонообмінним і УФ-опроміненням, розділяючи розчинні мікрометали на сім груп. На першому етапі анодна зачистка вольтамметрії в буфері оцтової кислоти pH 4.8 розрізняє лабільні метали та нелабільні метали. Лише лабільні метали - ті, що присутні у вигляді гідратованих іонів, слабозв'язаних комплексів або слабо адсорбованих на колоїдних поверхнях - осідають на електроді і дають сигнал. Загальні концентрації металів визначають по АСВ після перетравлення зразка в 2 М HNO 3 протягом 5 хв, що перетворює всі метали в ASV-лабільну форму.
Іонообмінна смола Chelex-100 додатково розрізняє сильно зв'язані метали - як правило, метали, пов'язані з неорганічними та органічними твердими речовинами, але також тісно пов'язаними з хелатними лігандами - і більш слабко зв'язаними металами. Нарешті, УФ-випромінювання розрізняє метали, пов'язані з органічними фазами, та неорганічними фазами. Аналіз зразків морської води, наприклад, свідчить про те, що кадмій, мідь та свинець присутні насамперед у вигляді лабільних органічних комплексів або лабільних адсорбатів на органічних колоїдах (група II в табл.\(\PageIndex{1}\)).
Диференціальна імпульсна полярографія та зачистка вольтамметрії використовуються для визначення мікрометалів у повітряно-дисперсних частинок, золі, породах, мінералах та відкладеннях. Сліди металів, звичайно, спочатку вносяться в розчин за допомогою травлення або екстракції.
Амперометричні датчики також використовуються для аналізу зразків навколишнього середовища. Наприклад, описаний раніше датчик розчиненого O 2 використовується для визначення рівня розчиненого кисню та біохімічної потреби в кисні, або БПК, вод і стічних вод. Останній тест - який є мірою кількості кисню, необхідного водним бактеріям, коли вони розкладають органічну речовину, є важливим при оцінці ефективності очисних споруд та моніторингу органічного забруднення природних вод. Високий БПК говорить про те, що вода має високу концентрацію органічних речовин. Розпад цієї органіки може серйозно виснажити рівень розчиненого кисню у воді, негативно впливаючи на водну життєдіяльність. Інші амперометричні датчики доступні для моніторингу аніонних поверхнево-активних речовин у воді та CO 2, H 2 SO 4 та NH 3 в атмосферних газах.
Клінічні зразки
Диференціальна пульсова полярографія і стріппінг-вольтамметрія використовуються для визначення концентрації мікрометалів в різних клінічних зразках, включаючи кров, сечу і тканини. Визначення свинцю в крові представляє чималий інтерес через побоювання з приводу отруєння свинцем. Оскільки концентрація свинцю в крові настільки мала, анодна зачистка вольтамметрії часто є більш підходящою методикою. Аналіз ускладнюється, однак, наявністю білків, які можуть адсорбуватися до ртутного електрода, гальмуючи або осадження, або зачистку свинцю. Крім того, білки можуть перешкоджати електроосадженню свинцю за рахунок утворення стабільних, нелабільних комплексів. Перетравлення та омивання зразка крові mini- усуває цю проблему. Диференціальна імпульсна полярографія корисна для рутинного кількісного аналізу лікарських засобів у біологічних рідинях, при концентраціях менше 10 —6 М [Brooks, M.A. «Застосування електрохімії до фармацевтичного аналізу», Глава 21 в Кіссінджер, П.Т.; Heinemann, W.R., ред. Лабораторні методи в електроаналітичній хімії, Марсель Деккер, Inc.: Нью-Йорк, 1984, стор. 539—568.]. Амперометричні датчики з використанням ферментних каталізаторів також мають безліч клінічних застосувань, кілька прикладів яких наведені в табл\(\PageIndex{2}\).
Різні зразки
Крім екологічних зразків і клінічних зразків, диференціальна імпульсна полярографія і зачистка вольтамметрія використовуються для аналізу мікрометалів в інших зразках, включаючи харчові продукти, сталі та інші сплави, бензин, залишки пороху, фармацевтичні препарати. Вольтамметрія є важливою методикою кількісного аналізу органіки, особливо у фармацевтичній промисловості, де вона використовується для визначення концентрації ліків і вітамінів у рецептурах. Наприклад, для кількісного аналізу вітаміну А, ніацинаміду та рибофлавіну доступні вольтамметричні методи. Коли сполука інтересу не є електроактивною, вона часто може бути виведена до електроактивної форми. Одним із прикладів є диференціальне імпульсне полярографічне визначення сульфаніламіду, який перетворюється в електроактивний азобарвник шляхом з'єднання з сульфаміновою кислотою і 1-нафтолом.
У попередньому розділі ми дізналися, як використовувати вольтамметрію для визначення концентрації аналіта в різних зразках. Ми також можемо використовувати вольтамметрію для характеристики властивостей аналіта, включаючи перевірку його електрохімічної оборотності, визначення кількості електронів, переданих під час його окислення або відновлення, та визначення його постійної рівноваги в зв'язаній хімічній реакції.
Характеристика додатків
У застосуванні характеристик ми вивчаємо властивості системи. Тут описано три приклади: визначення, чи є окислювально-відновна реакція електрохімічно оборотною, визначення кількості електронів, що беруть участь у окисно-відновній реакції, і вивчення комплексоутворення метал-лігандів.
Електрохімічна оборотність і визначення n
Раніше в цьому розділі ми вивели зв'язок між E 1/2 та потенціалом стандартного стану для окислювально-відновної пари, використовуючи рівняння Нернста, зазначивши, що окислювально-відновна реакція повинна бути електрохімічно оборотною. Як ми можемо визначити, чи зворотна окислювально-відновна реакція, дивлячись на її вольтаммограму? Як ми дізналися в розділі 25.3, для оборотної окислювально-відновної реакції зв'язок між потенціалом і струмом становить
\[E=E_{½} - \frac{0.05916}{n} \log \frac{i}{i_{l} - i} \label{app3} \]
Якщо реакція електрохімічно оборотна, то ділянка Е проти\(\log \frac{i}{i_l - i}\) є прямою лінією з ухилом —0,05916/ п. Крім того, нахил повинен давати ціле значення для n.
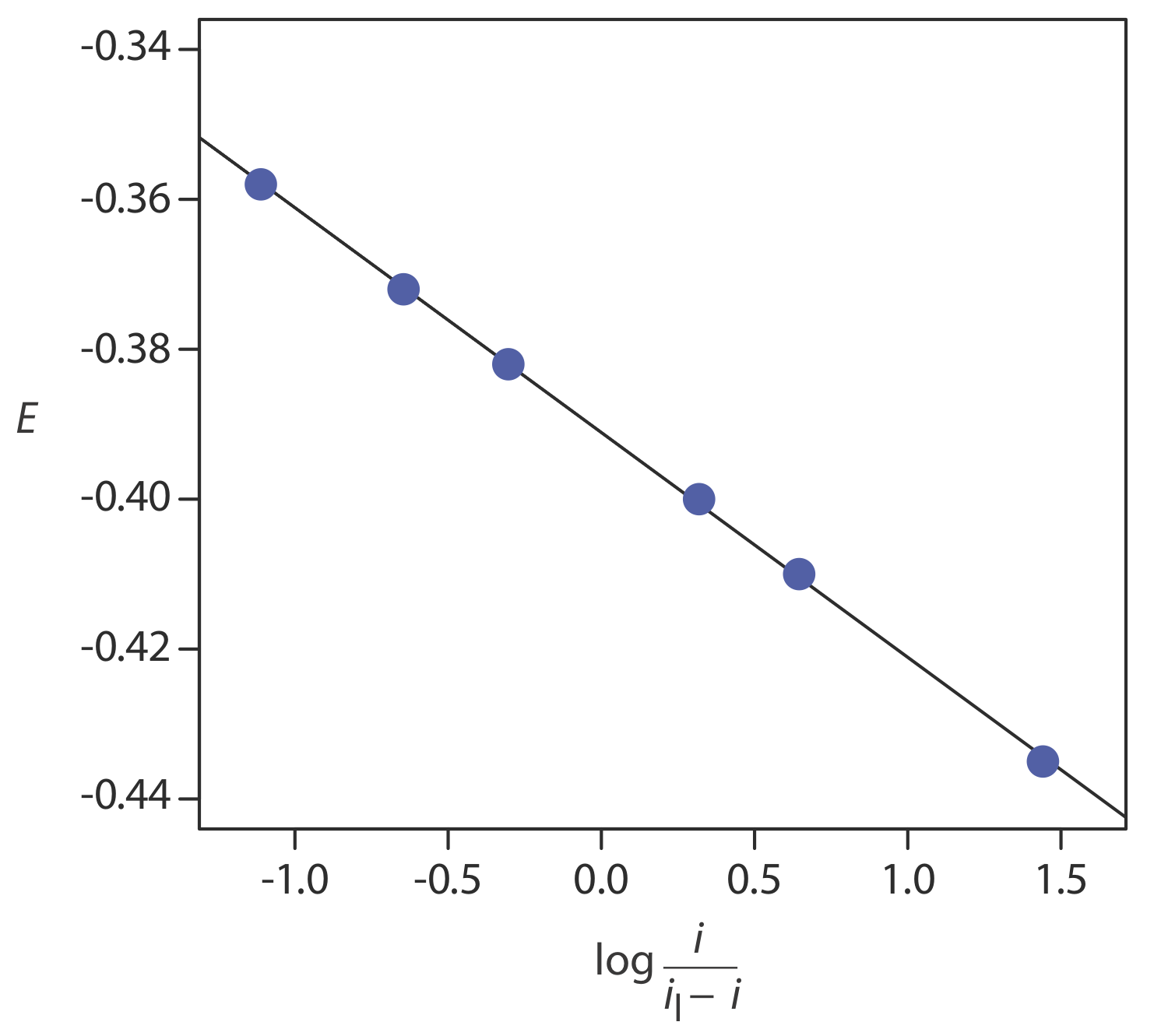
Наступні дані були отримані з лінійного сканування гідродинамічної вольтаммограми реверсивної реакції відновлення.
| E (V проти SCE) | струм (мкА) |
|---|---|
| —0,358 | 0,37 |
| —0,372 | 0,95 |
| —0,382 | 1.71 |
| —0,400 | 3.48 |
| —0,410 | 4.20 |
| —0,435 | 4.97 |
Граничний струм 5,15 мкА. Показати, що реакція відновлення оборотна, і визначити значення для n і для E 1/2.
Рішення
\(\PageIndex{3}\)На малюнку показаний сюжет Е проти\(\log \frac{i}{i_l - i}\). Оскільки результат є прямою, ми знаємо, що реакція є електрохімічно оборотною в умовах експерименту. Лінійний регресійний аналіз дає рівняння для прямої
\[E=-0.391 \mathrm{V}-0.0300 \log \frac{i}{i_{l}-i} \nonumber \]
З Рівняння\ ref {app3} нахил еквівалентний —0,05916/ n; розв'язування для n дає значення 1,97, або 2 електронів. З рівняння\ ref {app3} ми також знаємо, що E 1/2 є y -перехоплення для ділянки E проти\(\log \frac{i}{i_l - i}\); таким чином, E 1/2 для даних у цьому прикладі дорівнює —0.391 V проти SCE.
Ми також можемо використовувати циклічну вольтамметрію для оцінки електрохімічної оборотності, розглядаючи різницю між піковими потенціалами для анодного та катодного сканування. Для електрохімічно оборотної реакції відповідає таке рівняння.
\[\Delta E_{p}=E_{p, a}-E_{p, c}=\frac{0.05916 \ \mathrm{V}}{n} \label{app4} \]
Як приклад, для двоелектронного зменшення ми очікуємо a\(\Delta E_p\) приблизно 29,6 мВ. Для електрохімічно незворотної реакції значення більше\(\Delta E_p\), ніж очікувалося.
Визначення констант рівноваги для пов'язаних хімічних реакцій
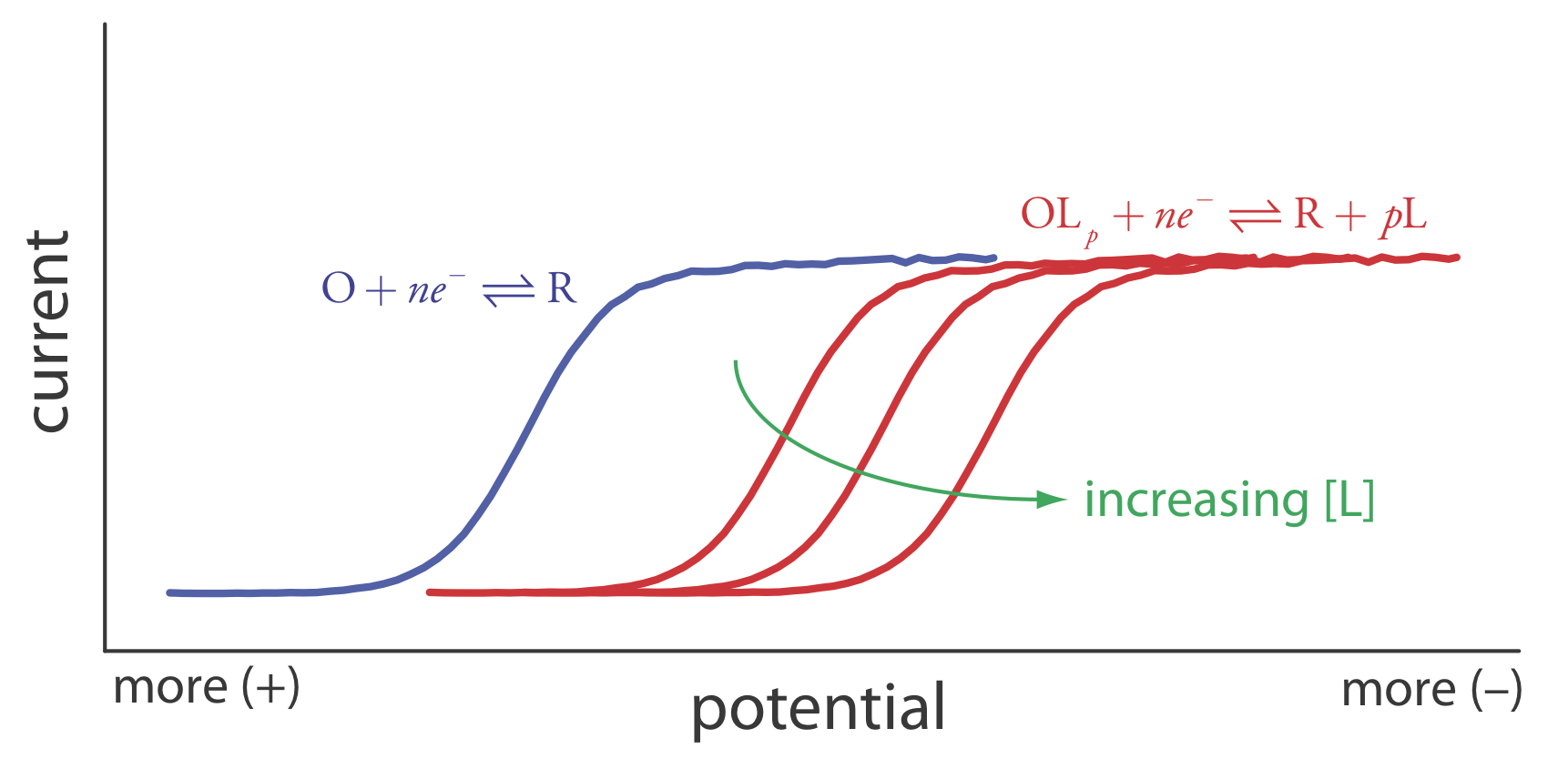
Іншим важливим застосуванням вольтамметрії є визначення постійної рівноваги для реакції розчину, яка пов'язана з окислювально-відновною реакцією. Наявність реакції розчину впливає на легкість перенесення електронів в окисно-відновної реакції, зміщуючи Е 1/2 в більш негативний або на більш позитивний потенціал. Розглянемо, наприклад, зменшення O до R
\[O+n e^{-} \rightleftharpoons R \label{app5} \]
вольтаммограма для якої показана на рис\(\PageIndex{4}\). Якщо ввести ліганд, L, який утворює сильний комплекс з О, то ми також повинні розглянути реакцію
\[O+p L\rightleftharpoons O L_{p} \label{app6} \]
При наявності ліганда загальна окислювально-відновна реакція становить
\[O L_{p}+n e^{-} \rightleftharpoons R+p L \label{app7} \]
Через його стабільності зниження комплексу ОЛ р менш сприятливо, ніж зниження О. Як показано на малюнку\(\PageIndex{4}\), отримана вольтаммограма зміщується до потенціалу, який є більш негативним, ніж для O. Крім того, зсув вольтаммограми збільшується в міру збільшення концентрації ліганду.
Ми можемо використовувати цей зсув у значенні E 1/2 для визначення як стехіометрії, так і константи формування для металолігандного комплексу. Щоб вивести зв'язок між відповідними змінними, ми починаємо з двох рівнянь: рівняння Нернста для зменшення O
\[E=E_{O / R}^{\circ}-\frac{0.05916}{n} \log \frac{[R]_{x=0}}{[O]_{x=0}} \label{app8} \]
і постійна стабільність,\(\beta_p\) для комплексу метал-ліганд на поверхні електрода.
\[\beta_{p} = \frac{\left[O L_p\right]_{x = 0}}{[O]_{x = 0}[L]_{x = 0}^p} \label{app9} \]
При відсутності ліганду напівхвильовий потенціал виникає, коли [R] x = 0 і [O] x = 0 рівні; таким чином, з рівняння Нернста ми маємо
\[\left(E_{1 / 2}\right)_{n c}=E_{O / R}^{\circ} \label{app10} \]
де індекс «nc» означає, що комплексу немає. Коли ліганд присутній, ми повинні враховувати його вплив на концентрацію О. Розв'язування рівняння\ ref {app9} для [O] x = 0 і підставляючи в рівняння\ ref {app8} дає
\[E=E_{O/R}^{\circ}-\frac{0.05916}{n} \log \frac{[R]_{x=0}[L]_{x=0}^{p} \beta_{p}}{\left[O L_{p}\right]_{x=0}} \label{app11} \]
Якщо константа освіти досить велика, така, що по суті всі О присутні у вигляді комплексу OL p, то\([R]_{x = 0}\) і\([OL_p]_{x = 0}\) рівні по півхвильовому потенціалу, а Equation\ ref {app11} спрощує
\[\left(E_{1 / 2}\right)_{c} = E_{O/R}^{\circ} - \frac{0.05916}{n} \log{} [L]_{x=0}^{p} \beta_{p} \label{app12} \]
де індекс «c» вказує на те, що комплекс присутній. Визначення\(\Delta E_{1/2}\) як
\[\triangle E_{1 / 2}=\left(E_{1 / 2}\right)_{c}-\left(E_{1 / 2}\right)_{n c} \label{app13} \]
і підставляючи Equation\ ref {app10} і Equation\ ref {app12} і розширення терміну журналу залишає нам наступне рівняння.
\[\Delta E_{1 / 2}=-\frac{0.05916}{n} \log \beta_{p}-\frac{0.05916 p}{n} \log {[L]} \label{app14} \]
Ділянка\(\Delta E_{1/2}\) проти колоди [L] являє собою пряму лінію, з нахилом, який є функцією стехіометричного коефіцієнта комплексу метал-лігандів, p, і y -перехоплення, що є функцією константи його формування\(\beta_p\).
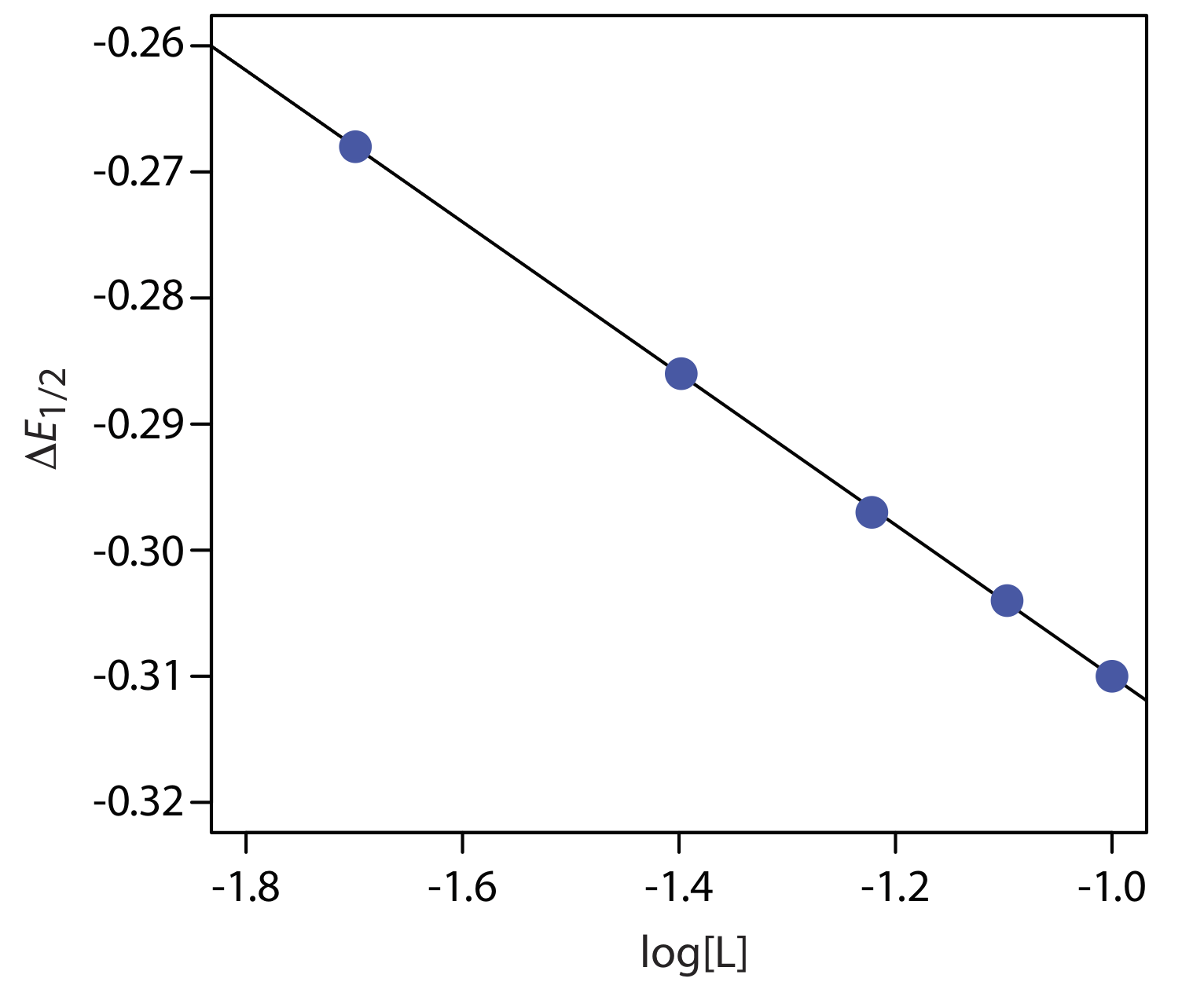
Вольтаммограма для двоелектронного відновлення (n = 2) металу, M, має півхвильовий потенціал —0,226 В проти SCE. При наявності надлишку ліганду, L реєструють наступні півхвильові потенціали.
| [Л] (М) | (E 1/2) c (V проти СКЕ) |
|---|---|
| 0,020 | —0,494 |
| 0,040 | —0.512 |
| 0,060 | —0.523 |
| 0,080 | —0,530 |
| 0.100 | —0.536 |
Визначають стехіометрію комплексу метал-ліганд і константу його формування.
Рішення
Почнемо з обчислення значень\(\Delta E_{1/2}\) за допомогою Equation\ ref {app13}, отримавши значення в наступній таблиці.
| [Л] (М) | \(\Delta E_{1/2}\)(V проти SCE) |
|---|---|
| 0,020 | \ (\ Дельта E_ {1/2}\) (V проти СКЕ) ">—0.268 |
| 0,040 | \ (\ Дельта E_ {1/2}\) (V проти СКЕ) ">—0.286 |
| 0,060 | \ (\ Дельта E_ {1/2}\) (V проти СКЕ) ">—0.297 |
| 0,080 | \ (\ Дельта E_ {1/2}\) (V проти СКЕ) ">—0.304 |
| 0.100 | \ (\ Дельта E_ {1/2}\) (V проти СКЕ) ">—0.310 |
\(\PageIndex{5}\)На малюнку показаний отриманий графік\(\Delta E_{1/2}\) як функція log [L]. Лінійний регресійний аналіз дає рівняння для прямої
\[\triangle E_{1 / 2}=-0.370 \mathrm{V}-0.0601 \log {[L]} \nonumber \]
З Рівняння\ ref {app14} ми знаємо, що нахил дорівнює —0.05916 p/n. Використовуючи ухил і n = 2, вирішуємо для p отримання значення 2,03 ≈ 2. Таким чином, стехіометрія комплексу становить ML 2. Ми також знаємо, з Рівняння\ ref {app14}, що y -перехоплення еквівалентно — (0.05916/ n) журналу\(\beta_p\). Рішення для\(\beta_2\) дає константу формування\(3.2 \times 10^{12}\).
Циклічна вольтамметрія є однією з найпотужніших електрохімічних методик дослідження механізму пов'язаних електрохімічних і хімічних реакцій. Лікування цього аспекту циклічної вольтамметрії виходить за межі цього тексту, хоча ви можете звернутися до додаткових ресурсів цієї глави для отримання додаткової інформації.