12.7: Хронічний мієлолейкоз (ХМЛ)
- Page ID
- 5173

Лейкемія - це неконтрольована проліферація одного виду лейкоцитів (або лейкоцитів). Як і всі онкологічні захворювання (напевно), все лейкозні клітини походять з однієї клітини, яка втратила здатність підтримувати нормальний контроль над клітинним циклом. Існує ряд типів лейкемії, як і слід було очікувати від кількості типів лейкоцитів (5) та кількості стадій, через які вони проходять у міру дозрівання. Одним з найпоширеніших є c hronic m yelogenic l eukemia або ХМЛ.
Хронічний мієлогенний лейкоз (ХМЛ) виникає в стовбуровій клітині кісткового мозку, яка є попередником всіх типів клітин крові. Однак він зазвичай вражає так звану мієлоїдну лінію (звідси і назва), яка виробляє гранулоцити і макрофаги. Як випливає з назви, захворювання часто існує роками лише з помірно підвищеною кількістю лейкемічних клітин (що походять від стовбурових клітин) і мало симптомів. Однак у якийсь момент пацієнт переживає «вибухову кризу», коли лейкемічні гранулоцитарно-макрофагові попередники починають ділитися самі по собі - надзвичайно збільшуючи їх кількість, не продовжуючи їх диференціацію.
Філадельфійська хромосома (Ph 1)
У більшості випадків ХМЛ лейкемічні клітини мають хромосомну аномалію, не виявлену ні в будь-яких нелейкемічних лейкоцитах, ні в будь-яких інших клітині організму пацієнта. Ця аномалія являє собою реципрокну транслокацію між однією хромосомою 9 і однією хромосомою 22. Ця транслокація позначається t (9; 22). Це призводить до того, що одна хромосома на 9 довше норми і одна хромосома на 22 коротша за норму. Остання називається філадельфійської хромосомою і позначається\(Ph^1\).
ДНК, видалена з хромосоми 9, містить більшу частину протоонкогена, позначеного C-abL. Розрив хромосоми 22 відбувається в середині гена, позначеного BCR. Отримана філадельфійська хромосома має 5' розділ BCR злитий з більшістю C-abL.
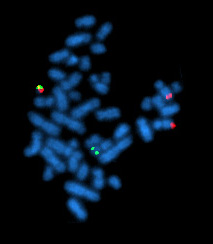
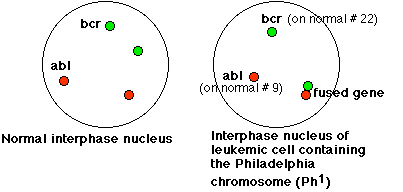
Мікрофотографія на малюнку\(\PageIndex{2}\) використовує флуоресценцію i n s itu h гібридизації (FISH) для виявлення ДНК ABL (червоний) та ДНК BCR (зелений) у міжфазних ядрах лейкемічних клітин пацієнта з ХМЛ. Червона точка в лівому центрі виявляє розташування ABL на нормальній хромосомі 9; зелена точка (верхній центр) показує BCR на нормальній хромосомі 22. Комбіновані точки (червоний + зелений = жовтий) внизу праворуч виявляють злитий ген BCR-ABL на хромосомі Філадельфії. Малюнок 12.7.3 являє собою схему, яка може допомогти вам інтерпретувати мікрофотографію.
Транскрипція та трансляція гібридного гена BCR-ABL виробляє аномальний («злитий») білок, який активує конституційно (весь час) ряд клітинних активностей, які зазвичай включаються лише тоді, коли клітина стимулюється фактором росту, таким як фактор росту тромбоцитів ( ФДГФ).
Ця нестримна активація збільшує швидкість мітозу і захищає клітину від апоптозу. Результатом є збільшення кількості Ph 1 -містять клітин. Під час хронічної фази захворювання вони все ще здатні вийти з клітинного циклу і диференціюватися на зрілі клітини, які виконують свої нормальні функції. Однак у якийсь момент в одній з цих клітин відбудеться інша мутація в протоонкогені (RAS, наприклад) або в гені супресора пухлини (p53, наприклад). Додаткова мутація спричиняє різке зростання швидкості мітозу в цій клітині та її нащадках. Дочірні клітини не диференціюються, і пацієнт вступає в кризову фазу захворювання.
До недавнього часу єдиним успішним лікуванням ХМЛ було знищення кісткового мозку пацієнта, а потім відновлення вироблення клітин крові шляхом вливання стовбурових клітин з кісткового мозку здорового донора. Але зараз лікування препаратом іматинібу мезилат (Gleevec® також відомий STI571), схоже, здатне вилікувати хворобу. Ця молекула вписується в активну ділянку білка ABL, запобігаючи АТФ зв'язуватися там. Без АТФ як донора фосфатів білок ABL не може фосфорилювати свій субстрат (и). Дослідження фази 2 показало, що майже 90% пацієнтів з ХМЛ, які лікувалися препаратом, не показали подальшого прогресування свого захворювання.
Gleevec також демонструє обіцянку проти одного типу раку шлунка (шлунково-кишкові стромальні пухлини = GIST), який є небезпечним для життя надмірним виробленням еозинофілів. При цьому захворюванні Глівек пригнічує різну гіперактивну тирозинкіназу. Цей також є результатом злиття частин двох різних генів (через видалення ДНК між ними):
- перші 233 кодони гена, позначеного FIP1L1, злиті з
- кінцеві 523 кодони гена (PDGFRα), що кодують домен тирозинкінази рецептора фактора росту тромбоцитів. Вироблений білок злиття, як і BCR-ABL, гіперактивний.