16.6: Морфометичний аналіз
- Page ID
- 6536

Морфометрія та фізичні маркери
Морфометрія (морфо — форма; метрики — вимірювання) — це використання фізичних вимірювань для визначення спорідненості організмів. З вимерлими організмами, які давно вимерли, екстракція ДНК виявляється складною. Аналогічним чином, до технологій ДНК для аналізу видів, таксономія Ліннея приписувалася організмам на основі подібності в особливостях.
Опис видів і варіації морфології
Нижче представлені зображення черепа орієнтирів сімейства ящірок Varanidae. До цього сімейства належать варани і дракони Комодо. Як видно нижче, загальна морфологія черепів досить схожа, щоб всі вони зберігали однакові орієнтири. На малюнку нижче також ілюструється різноманітність цих ящірок, які ілюструють велику різницю між видами.
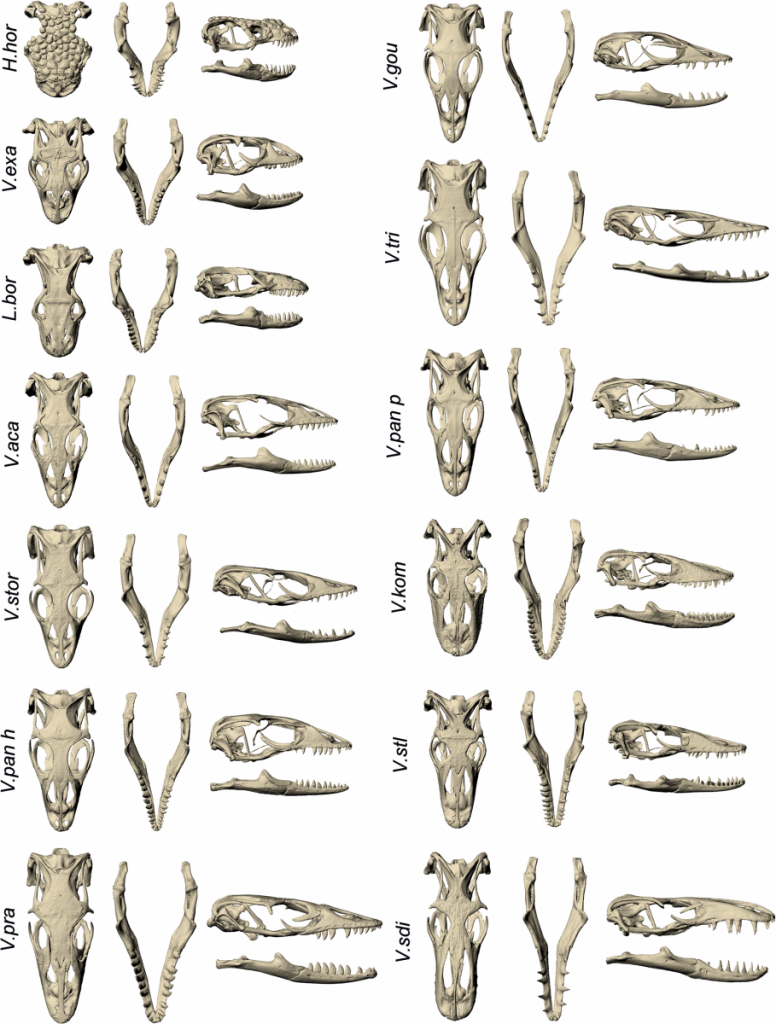
Черепа видів, що беруть участь в даному аналізі. Маккаррі та співавт. (2015) (CC-BY)
Орієнтири Стандартизувати вимірювання
Наявність набору спільних орієнтирів дає можливість проводити систематичні виміри морфометричних ознак.

Орієнтири та вимірювальні метрики для морфометричного аналізу черепів. Маккаррі та співавт. (2015) (CC-BY)
Евклідова відстань для вимірювання спорідненості
Евклідова відстань - це вимірювання, отримане з геометрії Піфагора, яке описує найкоротшу відстань (\(d\)) між 2 точками (\(A\)і\(B\)) як пряму лінію за допомогою тріангуляції. У декартовому просторі точки можуть бути визначені:
\[A = \left( x _ { A } , y _ { A } \right)\nonumber \]
і
\[B = \left( x _ { B } , y _ { B } \right)\nonumber \]
Стандартна теорема Піфагора може бути виражена у вигляді:
\[x ^ { 2 } + y ^ { 2 } = d ^ { 2 }\nonumber \]
Щоб знайти відстань між двома точками, ми використовуємо алгебру для обчислення .
.
\[d = \sqrt { x ^ { 2 } + y ^ { 2 } }\nonumber \]
У цьому випадку розгортаємо порівняння координат двох точок:
\[\Delta x = x _ { B } - x _ { A }\nonumber \]
і
\[\Delta y = y _ { B } - y _ { A }\nonumber \]

Потім ми можемо розширити цю ідею, включивши відмінності в точках даних, які описують порівняння декількох вимірювань.
\[d \left( \mathbf { X } _ { \mathbf { i } } , \mathbf { X } _ { \mathbf { j } } \right) = \sqrt { \sum _ { k = 1 } ^ { p } \left( X _ { i k } - X _ { j k } \right) ^ { 2 } }\nonumber \]
Обчислення відстані за допомогою R

- Завантажте набір даних (McCurry et al. 2015), пов'язаного з цією діяльністю (файл CSV, відокремлених комами). Це може бути використано в електронній таблиці або в текстовому редакторі. Ці дані можна імпортувати в R для визначення евклідових відстаней орієнтирів.
- Наступний код у R завантажить набір даних у змінну під назвою «varanoid», виміряє евклідову відстань та збереже графік у PDF-файл у каталозі під назвою «/tmp».
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 |
|
Аналіз ДНК
Перш ніж почати цю діяльність, перегляньте біоінформатику та аналіз послідовностей.
- Пошук NCBI для мітохондріальних послідовностей від видів, що беруть участь у McCurry 2015. Дані були представлені компанією Аст (2001).
- Знайдіть послідовності та ідентифікувати/витягуйте елементи, які є загальними для всіх.
- Зберіть спільні послідовності в текстовому редакторі як єдиний файл FASTA, де кожен вид розділений заголовком («>Види А»).
- Блокнот на Windows (але краще завантажити notepad++)
- Textedit на Mac (але, ймовірно, краще завантажити TextWrangler)
- Гедіт на Лінукс
- Збережіть файл як «something.fasta».
- Виконайте аналіз декількох послідовностей за допомогою UGENE.
- Створіть філогенетичне дерево за допомогою UGENE. Для цієї вправи використовуйте максимальну правдоподібність (PhyML) як алгоритм. Файл підручник нижче.
- Порівняйте ДНК з морфометричними аналізами. Які проблеми ми могли собі уявити, виникають, якщо покладатися виключно на морфометрію?
Посилання
- McCurry MR, Mahony M, Клаузен PD, Quayle MR, Уолмслі CW, Джессоп Т.С., Wroe S, Річардс H, МакГенрі КР. (2015) Взаємозв'язок між черепною структурою, Біомеханічні показники та екологічне різноманіття у вараноїдних ящірок. Лос Один 10 (6): e0130625. Номер користувача: 10.1371/журнальний телефону.0130625
- Аст, Дженніфер С. (2001) Докази та еволюція мітохондріальної ДНК у Вараноідеї (Squamata). Класистика 17 (3): 211—26. http://www.sciencedirect.com/science/article/pii/S0748300701901690
- Фішер, Р.А. (1936) Використання множинних вимірювань в таксономічних задачах. Літопис Євгеніки, 7:179—188. дої:10.1111/j.1469-1809.1936.tb02137.x