13: Синтез вітаміну B₂
- Page ID
- 24265
Загальний синтез вітаміну В 12 був здійснений у 1973 році завдяки грандіозному співробітництву між групою Р.Б. Вудворда в Гарвардському університеті (США) та групою А. Ешенмозера, Швейцарський федеральний технологічний інститут (ETH), Цюрих, Швейцарія. На виконання цього гігантського завдання знадобилося близько дванадцяти років і більше двох десятків старших вчених. Досягнення по-різному хвалять хіміки-органіки — монументальне досягнення в літописах органічної синтетичної хімії; прорив; миля каменю в органічному синтезі; неперевершене навіть через 40 років; завдання не менш авантюрне, ніж узгодження Евересту. На той час, коли почалася пригода, це був найгрізніший виклик у синтетичній хімії, на який мало хто наважився б. Оголошення про його синтез ознаменувало настання віку синтетичної органічної хімії. Вудворд і Ешенмозер працювали в тісній співпраці та в конкуренції в цьому історичному темпі. У процесі вони досягли не тільки вражаючого синтезу, а й відкрили кілька нових родовищ для майбутніх досліджень. Правило Вудворда Гофмана є найвідомішим з відгалужень. Вивчаючи цей синтез, студент повинен також задуматися над ретельністю у своєму плануванні всіх аспектів схеми та ініціювання відповідних базових досліджень заздалегідь, щоб полегшити основну схему на відповідних моментах. Це довге вступ якраз співзвучно довжині схеми, витраченим часом і чудовими досягненнями.
Ретроаналіз\(B_{12}\) синтезу вітамінів
На малюнку 13.1 показана будова Vit B 1 2 та основні структурні особливості/виклики складної молекули.
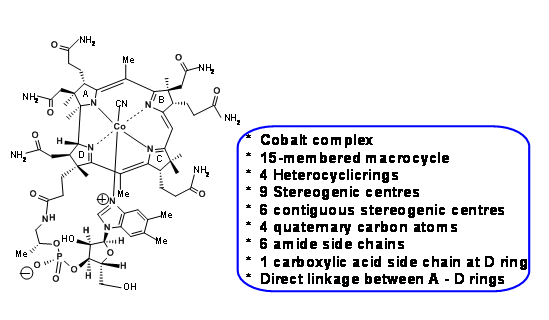
У своєму неповторному стилі Р.Б. Вудворд звертає увагу на ці виклики на початку своїх лекцій і на те, що для встановлення структури Vit B 1 2 шляхом хімічної деградації і, нарешті, рентгенівських дифракційних досліджень Дороті знадобилося близько 50 років Ходжкіна в 1956 році. Слід зазначити, що більшість досліджень хімічної деградації, якими б примітними вони не були, відбувалися в перші роки ХХ століття, коли сучасна органічна хімія перебувала в її створенні. Спектроскопічні дані, якщо такі були, були помітні через їх відсутність, і більшість деградації та синтетичної хімії були занадто суворими за сучасними (тобто 1960-х) стандартами для цієї делікатної молекули. Отже, це був складний сценарій. Це означало, що багато синтетичної хімії довелося заново винаходити, щоб задовольнити ці проблеми. Такі аспекти були ретельно сплановані, а плани на випадок надзвичайних ситуацій були розроблені заздалегідь. Вибагливі студенти могли побачити проблиск цього планування в цій короткій презентації. Ідеї з ретроаналізу, як ми розуміємо сьогодні, перебували в стадії розробки протягом шістдесятих років. Ретроаналізи, які ми використовуємо тут, є доповненнями інших вчених пізніше на основі фактів (лекцій, статей тощо), опублікованих двома групами. Всі такі цитати включаються в кінці написання.
Синтетична мішень була ідентифікована як Cobyric Acid, оскільки ця сполука була натуральним продуктом і була перетворена на Vit B 1 2 Bernhauer K., et.al., (1960) (рис. 13.2). Отже, загальний синтез кобірової кислоти становитиме формальний синтез Vit B 1 2.
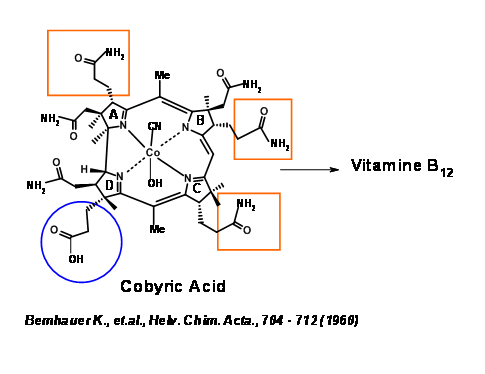
Кобірова кислота мала сім бічних ланцюгів карбонової кислоти, з яких чотири складалися фрагменти пропіонової кислоти, по одному на кожному гетероциклічному кільці. Основним завданням було диференціювати ланцюг пропіонової кислоти на кільці D від інших фрагментів оцтової кислоти та пропіонової кислоти. Тому було вирішено, що цей непарний кислотний фрагмент буде маскуватися під нітрил (рис. 13.3). Це все ще залишає складне завдання диференціації, яке ми могли б вирішити пізніше. Спочатку було вирішено розглядати молекулу як складену з двох половинок — Східної та Західної. Перше відключення було на кільцевому з'єднанні A/B на метиленовому мосту (1.13.3A). Розщеплення другого мосту на кільцевих переходах C/D дало східній половині як тіодекстролін, відповідальний за групу Ешенмозера в Цюріху та західній половині як ціанобромід, відповідальний за групу Вудворда в Гарварді (США).
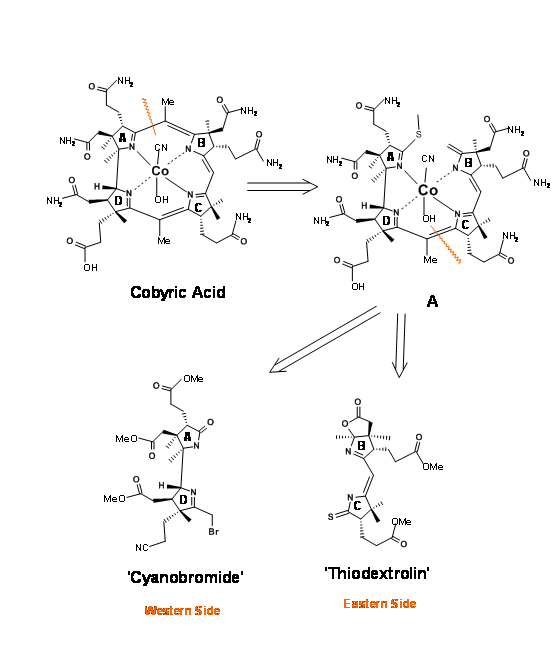
Ретроаналіз ціаноброміду
Ця західна половина має грізний масив з шести суміжних стереоцентрів на восьмивуглецевому каркасі. Зауважимо, що стереоцентри були заплановані на основі відомих стереоселективностей, а шість членних кілець були побудовані для забезпечення ланцюгів пропіонової кислоти (рис. 13.4). Азот для кільця А надходив з індолу, чиє бензольне кільце давало бічні ланцюги для кільця А. Кільцевий азот D прийшов через перестановку Бекмана. Корнорстерон (1.13.4A) був ключовим проміжним (кутовий камінь), який утримував усі стереоцентри та ланцюги на Західній половині.
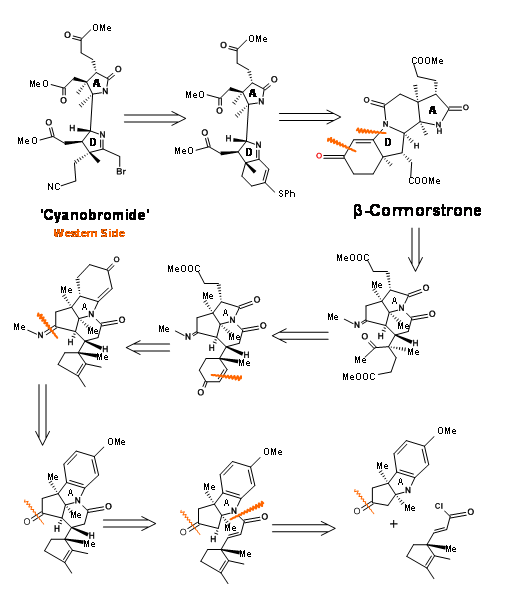
Синтез західної половини
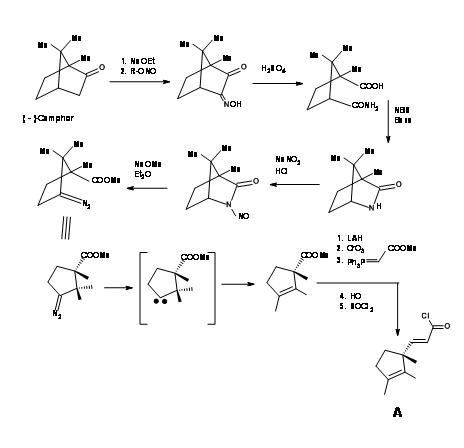
Необхідна одиниця енантіопура 1,2,3-триметиліклопентену надходила з камфорхінону, як показано на малюнку 13.5.
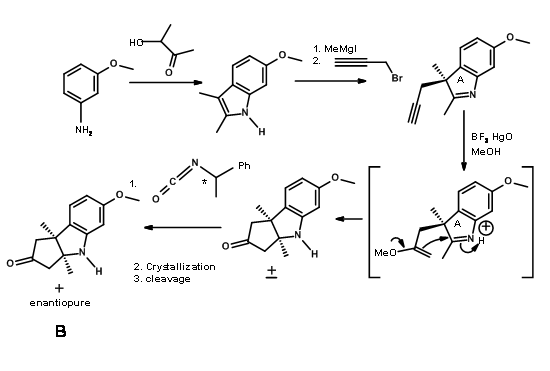
Через інший конвергентний синтез п'ятичленне кільце було сплавлено з індолом у зв'язку C2 - C3 і вирішено, як показано на малюнку 13.6.
(+) - енантіомер був фактично необхідний для цільового синтезу. Марний (-) - ентіомер був використаний як модельне з'єднання (для цього було «майже єдиним видом модельного дослідження, яке ми вважаємо цілком надійним» - RBW).
Фрагменти А і В були об'єднані, а потім оброблені до корнорстерону, як показано на малюнку 13.7.
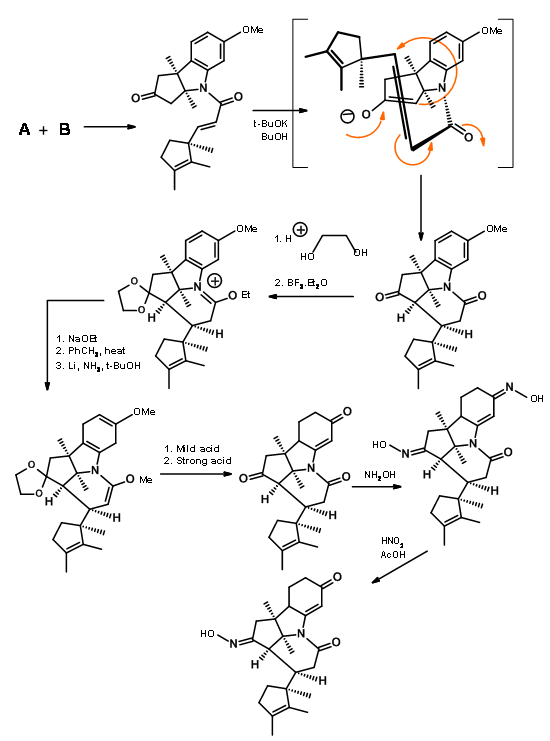
Зверніть увагу, що процес розширення кільця Бекмана привів в рух каскад реакції, що призводить до конденсації Клейзена та утворення кільця D, все в один етап. Шість членів іміду карбонілу також був розщеплений і поміщений ацетатний ланцюг на кільце D. Незважаючи на таке детальне планування та виконання, процес дав суміш епімерів у бічному ланцюзі пропіонової кислоти на кільці А, необхідний ізомер є незначним компонентом суміші. Основний небажаний продукт не розщеплюється на амідній зв'язку через несприятливого стеричного стиснення на бокових ланцюгах, що розвиваються (рис. 13.8).
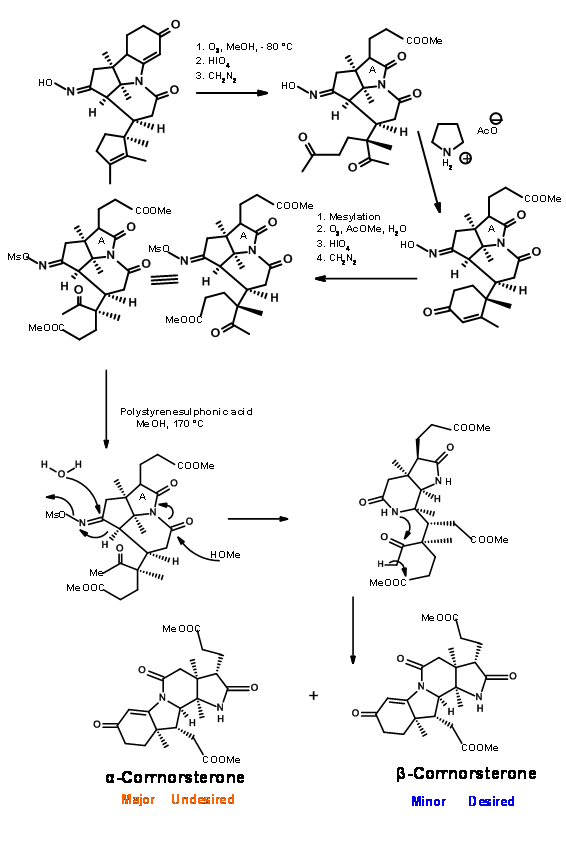
Однак ця несприятлива стерична проблема була вирішена незабаром. При гідролізі в сильних базових умовах амідне кільце відкрилося, а бічний ланцюг пропіонової кислоти ізомеризувався до менш напруженого ізомеру. Потім це може бути підкислений і етерифікований до β-корнорстерону з відновленням 90% бажаного ізомеру (рис. 13.9).
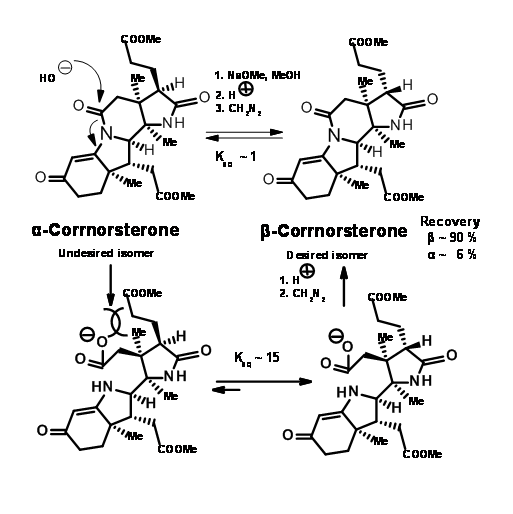
Цей правильний ізомер обробляли сумішшю метанолу і тіофенолу в кислотних умовах (рис. 13.10). Це призвело два процеси в рух. Тіофенол атакував кетон і активував цей центр, тоді як кисень метанолу атакував амідний зв'язок, що призводить до ефіру та ефіру тіоенолу.
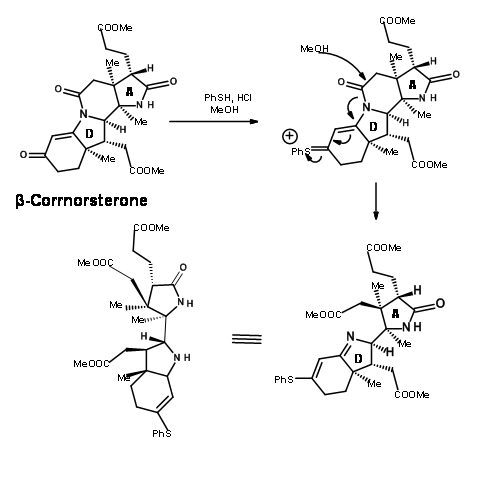
Озоноліз ефіру тіоенолу при -90°С розщепив олефінову одиницю до альдегід-тіоефірної сполуки (рис. 13.11). Тут розвивалася цікава нова хімія. Хоча тіоефіри менш реактивні до кислотного гідролізу і показали порівнянну реакційну здатність з кисневими нуклеофілами, азотні нуклеофіли були унікальними. Тіоефір реагував набагато швидше, ніж звичайні оксиефіри, щоб дати аміди. Таким чином, тіоефір був виключно розщеплений до аміду з аміаком, залишивши три метилових ефіри недоторканими. Потім фрагмент альдегіду вибірково перетворювали на спирт, а потім мезилювали в змішаних умовах ангідриду/піридину. Ця послідовність також перетворила амідний фрагмент в нітрил. Потім мезилат перетворювали в бромід, щоб дати ключовий проміжний ціанобромід.
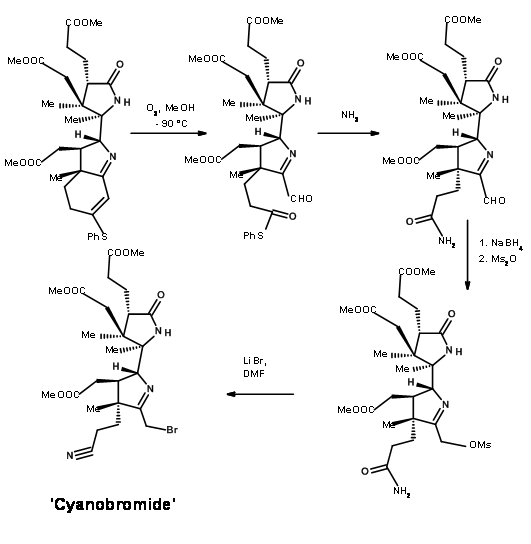
Цюріхська група одночасно працювала над синтезом східної половини під назвою тіодекстролін. Фрагменти для кілець B і C планувалися через., єдиний проміжний (1.13.12).
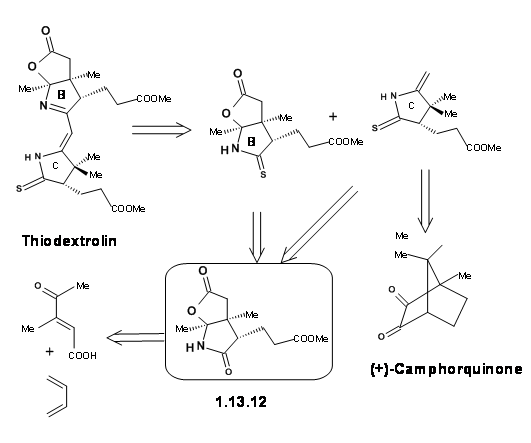
Синтез розпочався з реакції Дільса-Альдера, щоб правильно закріпити два асиметричні центри, і рацемат був вирішений. Чистий енантіомер дотримувався схеми для отримання сегмента кільця В. Такий же проміжний вийшов і фрагмент кільця С (рис. 13.13).
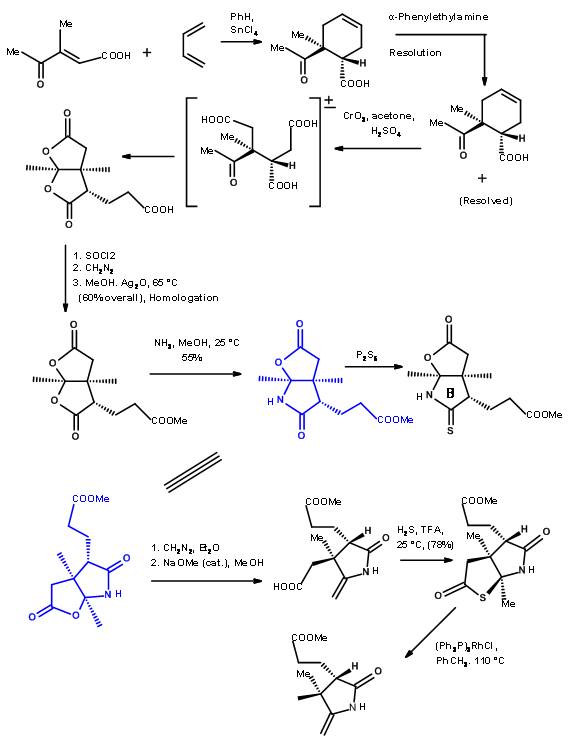
Для з'єднання двох таких фрагментів Ешенмозер розробив дві процедури сульфідного скорочення. Механізми процесів наведені нижче.
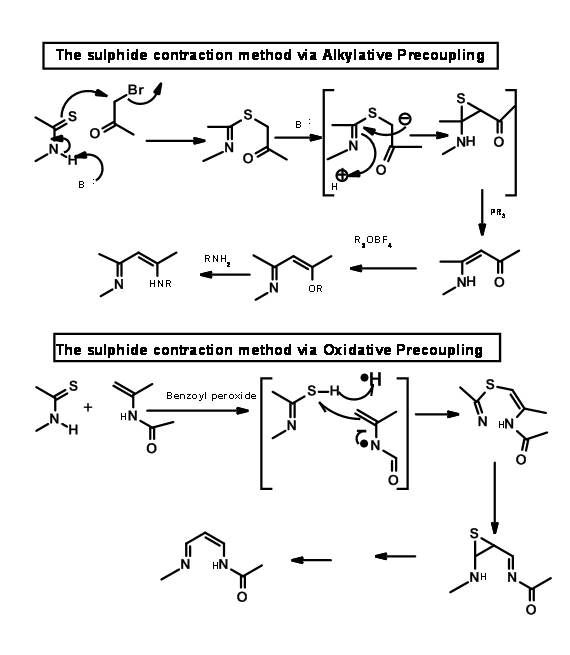
Використовуючи процедуру окислювального зчеплення/екструзії сірки, вони з'єднали кільця B і C, як показано на малюнку 13.15.
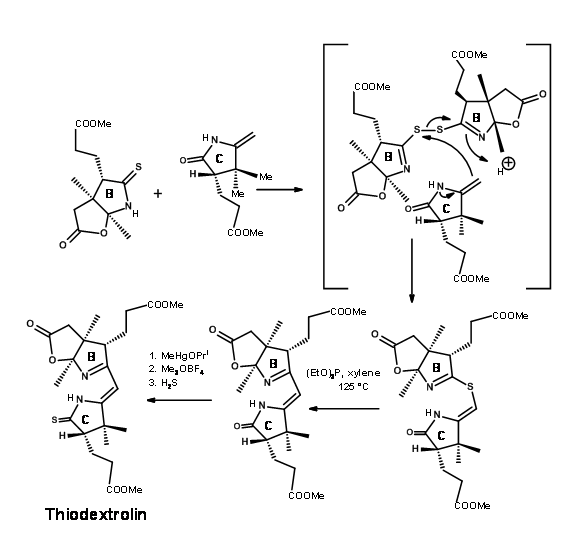
Група Вудворда також розробила новий синтез для кільця С, починаючи з (+) -Камфорхінону. Схема показана на малюнку 13.16.
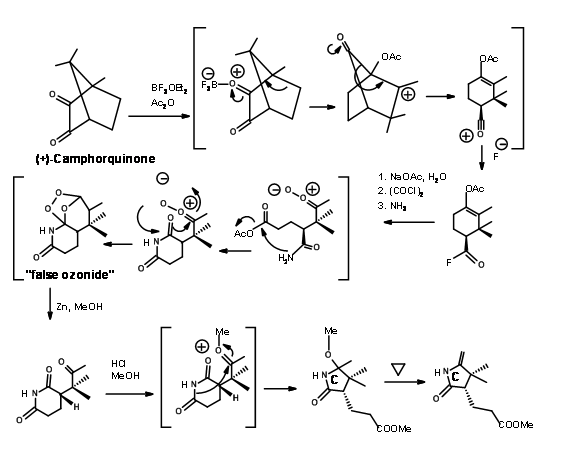
Хоча велика обережність була позбавлена стереоцентрів у всіх точках, проблеми стереоізомерів уникнути не вдалося. Синтезований кристалічний тіодекстролін був фактично сумішшю двох стереоізомерів у фрагменті пропіонової кислоти В-кільця. Хоча вони очистили суміш у цей момент, це не мало значення, оскільки цей стереоцентр був обумовлений подальшими порушеннями на пізніх стадіях. Суміш була взята вперед для першого зчеплення на мосту C/D. Після значних зусиль, які тривали більше року, вони були вперше з'єднані на південному кінці за допомогою процедури алкілування/екструзії (рис. 13.17). Зверніть увагу, що першим продуктом алкілування був тіоефір I типу, який легко ізомеризувався до тіоефіру II типу. Продукт отримав назву Ціанокоригенолід. Ця ізомеризація порушила стереоцентр на кільці С.
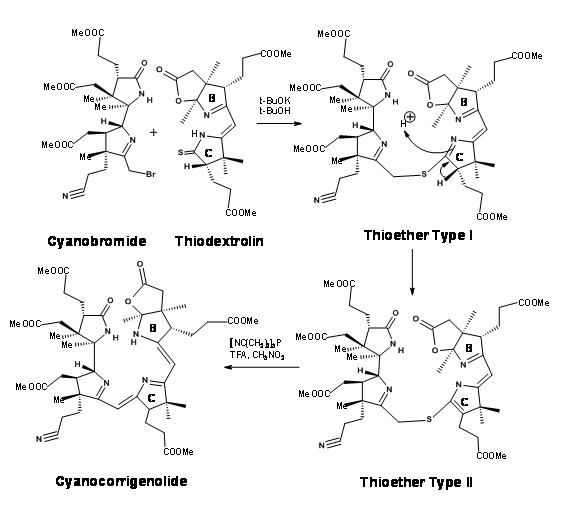
Наступним етапом стало формування моста А/Б. С/D з'єднане з'єднання спочатку обробляли фосфорним пентасульфідом з подальшим триметилоксонієм фторборатом (M e 3 OB F 4) (рис. 13.18). Ця процедура замінила кисень на кільцях A і B сіркою і, нарешті, на S-метилове похідне. Диметиламін в метнолі розщеплював тіолактонове кільце вибірково до диметилацетамідного ланцюга і кінцевого олефіну. Олефін був досить нестійким. Це повинно було бути негайно перетворено в кобальтовий комплекс. Ця процедура виявилася непростою. За кількох умов іон металу кобальту каталізував подальші реакції, що призводять до великого «руйнування» з'єднання. Після кількох експериментів було помічено, що хлорид кобальту або йодид в THF був унікальним для гладкого кобальтування. Процес комплексоутворення привів кільця A і B в безпосередній близькості. Базова каталізована реакція тоді дозволила утворенню мосту та видаленню сірчаного фрагмента найкращого стану, що каталізується циклізація DBN.
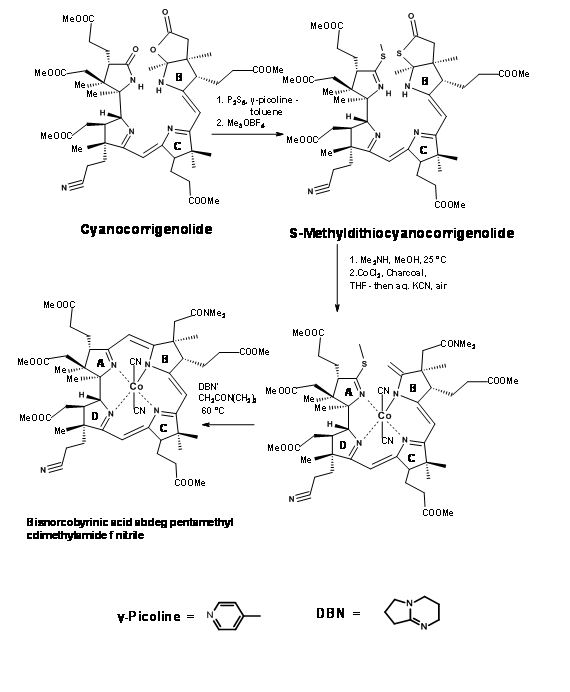
Зверніть увагу, що загальному перетворенню заважав асиметричний центр у кільця С. Тим не менш, міст A/B нарешті був на місці.
Цюріхська група також придумала альтернативну ZN-комплексну процедуру за тими ж лініями. Всі ці маніпуляції дійсно були жорсткими до (трьох) епімерізіруемих ланцюгів пропіонової кислоти на кільцях A, B і C. Кінцевий продукт був очищений TLC («пластинчаста хроматографія») і критично проаналізований ВЕРХ (новий хроматографічний інструмент на той час). УФ хромофори у всіх продуктах надали велику допомогу в цій хроматографії.
Зараз синтез має дві основні віхи. У молекулі є три активних метінових містка. Міст на кільцях В/С повинен був бути «захищений» від метилювання. Це було досягнуто шляхом окислювальної реакції утворення лактону на кільці В (I2, AcoH) (рис. 13.19). Цей новий четвертинний центр та існуючий четвертинний вуглець на C12 разом мали стеричний затори навколо C10. Хлорометиловий ефір увійшов виключно до центрів C5 та C15. Гідрогеноліз Ra-Ni розщепив тіоефіри і лактонове кільце в один прийом. За цим кроком послідувала етерифікація.
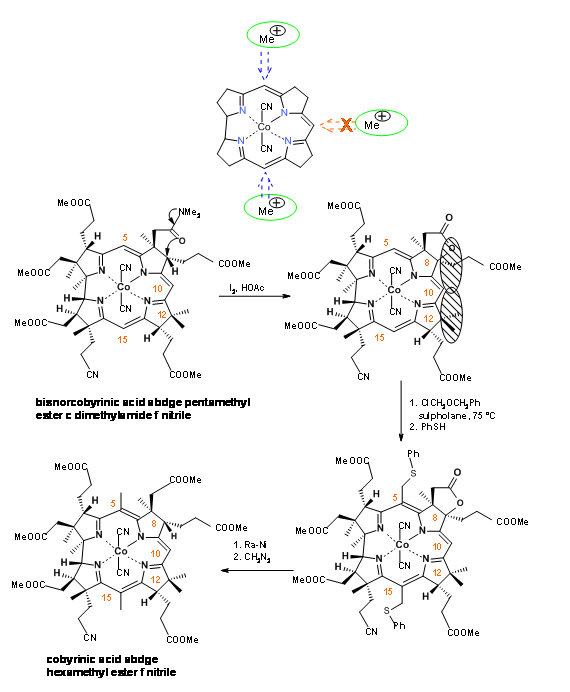
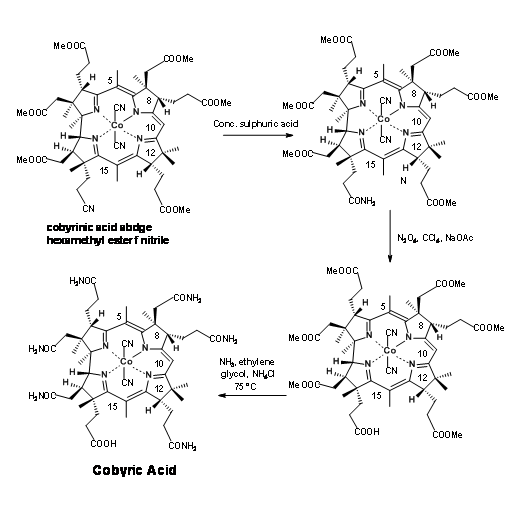
Конк. сірчана кислота перетворила нітрил в амід (рис. 13.20) .На цьому етапі перед ними постало складне завдання селективного розщеплення аміду, в присутності шести ефірів в молекулі. Після великих паралельних експериментів Гарвардська група знову відкрила ефективне селективне розщеплення амідного фрагмента в реагенті N 2 O 2.
Цюріхська група також вийшла з «диявольською схемою розщеплювача» для селективного гідролізу амідної групи в присутності ефірних груп. Дана схема показана на малюнку 13.21. Однак першій вважали за краще завдяки своїй простоті і кращої врожайності. Тим не менш, рішення Ешенмозера є свідченням людської винахідливості.
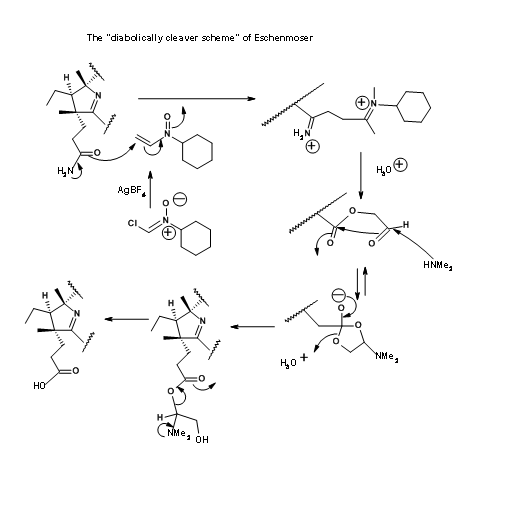
Останній крок цього довгого синтезу поставив серйозну проблему, яка потребує особливої згадки. Амітація ефірів аміаком була єдиним кроком, що залишився для остаточного нападу на синтез Кобірової кислоти. Більш уважний погляд показує, що завдання може бути не таким легким. Складні частини ефіру (зокрема, ацетатні одиниці) знаходилися в багатолюдних середовищах. Паралельно з цими розробками проводилися модельні дослідження на дуже схожих молекулах. На підставі цих досліджень, коли кислота гексаметилестеру 1 оброблялася аміаком в етиленгліколі при 75° C протягом 30 годин, отриманий продукт представляв собою не кобірову кислоту, а псевдокобірову кислоту, структура якої була встановлена як дегідрокобірова кислота. Цей продукт можна було очищати тільки за допомогою ВЕРХ. Це було одне з продуктів, отриманих раніше працівниками. Але про таке ускладнення вони поняття не мали. Ця таємниця зайняла кілька критичних досліджень, щоб розгадати. Протягом усіх цих досліджень була здійснена велика обережність, щоб побачити, що розчинники були добре дезоксигенізовані перед використанням. Кисень суворо уникали з реакційної атмосфери. Джерело цієї окислювальної циклізації приписували кобальту, який, як підозрювали, окислювально зв'язується з положенням С9, полегшуючи циклізацію амідного аніону. Ця небажана реакція зайняла багато часу, щоб вирішити. Кілька досліджень були спрямовані на амоніаліз в відновлювальних умовах, а також на відкриття лактамного кільця в умовах відновлення.
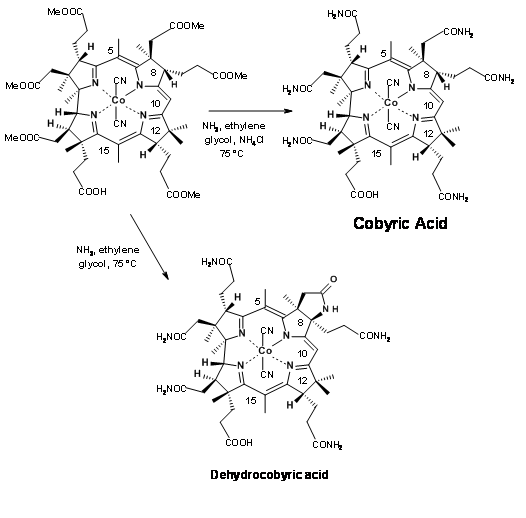
Нарешті кілька міліграмів ацетату амонію були додані до реакційної суміші, щоб запобігти утворенню амідного аніону, який, як підозрювали, є винуватцем циклізації (рис. 13.22). Цей трюк допоміг. Реакція була завершена протягом 10 годин, при цьому кобірова кислота була єдиним продуктом з хорошими врожаями. Ця кобірова кислота була ідентична за всіма параметрами, особливо у ВЕРХ, з натуральним продуктом, тим самим закінчивши цей довгий шлях для формального синтезу вітаміну В 12.
Це незвичайне пригода в органічному синтезі відзначається кількома найбільш значущими досягненнями.
- Розробки в хімії Corrin
- Синтетична стратегія
- Розробка декількох нових методологій
- Правила Вудворда-Гофмана
Навіть через півстоліття цей синтез Vit B 12 залишається неперевершеним і продовжує надихати покоління хіміків.
Подальше читання
- Останні досягнення в хімії натуральних продуктів, Woodward, R.B Pure & Appl. Хім., 17, 519 (1968).
- Ешенмозер, А. Чисте яблуко. Хім., 297—316 (1963).
- Ешенмозер, Анжев А.А. Хім. Int. Ред., 5 (1988)
- Останні досягнення в хімії натуральних продуктів, Woodward, R.B Pure & Appl. Хім., 25, 283 (1971).
- Загальний синтез вітаміну B 1 2, R. B. Woodward, Pure & Appl. Хім. , 33, 145 (1973).
- Синтез природного продукту та вітамін B 1 2, Eschenmoser, A; Wintner, C.E. Science, 196, 1410 (1977).
- Кроуфут-Ходжкін, Д. та ін. Природа, 178, 64 (1956).
- Фрідріх, В., ред., Вальтер де Груйтер: Берлін, 37, (1979).
- Цілі, Стратегії, Методи. Вайнгайм, Німеччина: VCH, (1996).
- Мистецтво і наука тотального синтезу на зорі ХХI століття, K.C. Nicolaou, Діонісіос Вурлуміс, Ніколас Вінссінгер, і Філ Баран, Angew. Хім. Int. Ред., 39, 44 (2000).
- Вітамін B 12: Епічна пригода в загальному синтезі, Ніл Гарг, 29 січня 2002 року.
- Асиметричний загальний синтез вітаміну В 1 2, Натан Вернер, зустріч групи Данії, 28 вересня 2010 р.
- Ніколау, К.С.; Соренсен, Е. J. Вітамін B 1 2. Класика в тотальному синтезі, ВЧ: Нью-Йорк, 100, (2003).