12: Синтез хлорофілу Вудворда
- Page ID
- 24351
Хлорофіл, найбільш помітний з натуральних продуктів, вже більше двох століть захоплюється хіміками-органіками не тільки своєю складною структурою, але і за його фотохімічний рулон у виробництві їжі, яка підтримує все живе на цій планеті. Ізоляційні та структурні роботи розпочалися наприкінці ХІХ ст. Ця монументальна діяльність, яка почалася з Willstätter, завершилася з'ясуванням повної структури хлорофілу «а» лише до середини ХХ століття. Більшість цих видатних творів, розтягнутих на півстоліття, мали місце, коли сучасна органічна хімія перебувала в зародковому стані. Синтез хлорофілу «а» Р.Б. Вудворда 1 визнаний видатним досягненням в органічному синтезі і входить до числа блискучих дорогоцінних каменів в синтезі. Попередній аналіз для синтезу, наполегливе заплановане приєднання до цієї складної, делікатної хімії його школи та логіка знаменитого Вудвардіанського підходу - все це хороші уроки для будь-якого вимогливого учня органічного синтезу.
Аналіз перед початком синтезу
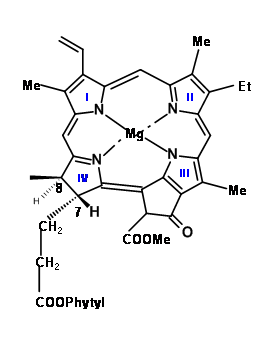
У цитованій лекції Вудворд починає свої дискусії про синтез з низки питань, які окреслюють не тільки критичні особливості структури під прикріпленням, але і приходить до правдоподібної мети і підходу до синтезу цієї складної молекули. Будова хлорофілу 'α' показано на (рис. 12.1).
Раніше робота встановила, що атом магнію та фітилову групу легко видалити та ввести на місце завдяки добре встановленій хімії. Хлорофіл 'α' належить до сімейства пігментів зеленого кольору хлори (12,2). Прості хлори легко окислюються (втрачають свої «зайві» водні), щоб дати більше кон'югованих порфіринів (12,3). Але деякі хлори, такі як хлорофіл, роблять це лише в жорстких умовах. Друга особливість, яка привернула їх увагу, - карбоциклічне п'ятичленне плавлене кільце, прикріплене до кільця III. Уважне вивчення молекулярних моделей припустило, що порфірин, який мав низку замісників на позиціях C5, C6, Cγ, C7 та C8, буде дуже переповнений, як показано в (12.3) та (12.4). Вони відзначили, що хлори і порфірини, які мали заміщення на Cγ і карбоксильну групу при С6, пухкі C O 2 з легкістю, при цьому відсутність Cγ заміни
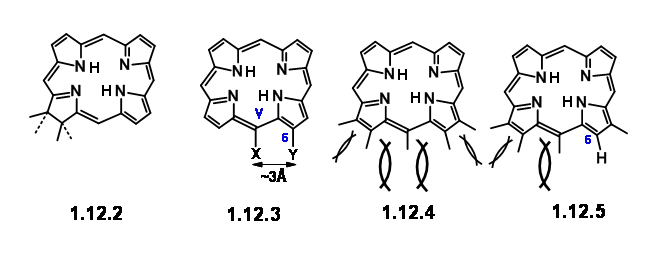
любив C6 карбонілу підвищеної стабільності. Всі ці особливості привели їх до висновку, що у порфіринів простір при C7/Cγ і Cγ /C6 найбільш переповнений. Декарбоксилювання зменшує цей штам частково (12,5). Наявність замісника при Cγ також збільшує деформацію простору C7/ Cγ. Ця скупченість, можливо, частково полегшується утворенням карбоциклічного кільця при Cγ /C6. Крім того, деформація при C7/Cγ може бути знята шляхом введення «додаткового» водню при C7 та C8. Ті ж фактори деформації впливали б на транс-розташування замісників при С7 і С8. Такий детальний аналіз даної структури не тільки допоміг їм зрозуміти дану структуру, але й припустив, що «Цільовим» для синтезу може бути порфілін, подібний до (12.6). Після його синтезу таку структуру можна змусити рухатися далі, щоб зайняти «зайві» водні та перейти до (12.7). Залишилися деякі питання. Вінілова група на першому кільці вважалася дуже чутливою, щоб протистояти строгості проектованого синтезу.
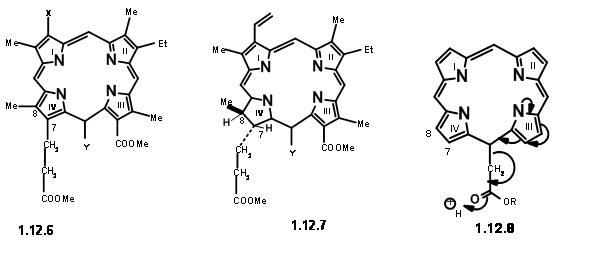
Значить, він був замінений еквівалентним амінопропановим ланцюгом. Як показано в структурі (12,7), залишок, необхідний при Cγ, є оцтовою кислотою. Однак механістичний аналіз такого порфірину (12.8) припустив, що така одиниця легко усуне через очікуваний «електронний фактор» (насправді зараз ми б сказали «емальоподібна активність»). Вони вирішили зробити цей агрегат залишком пропіонової кислоти, щоб уникнути цієї нестабільності. Зараз ми прибули до цільової структури (12,9). Грунтуючись на відомій хімії пірролу, вони вирішили зробити праву руку і ліву сторону молекули незалежно і конденсувати ці одиниці в тетрамер. Таким чином, вони прибули до наступних чотирьох мономерних одиниць (12.10).
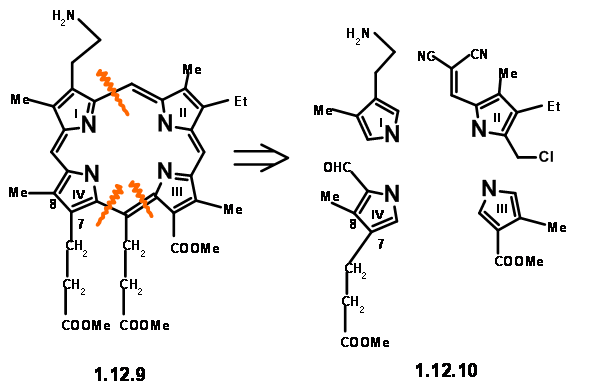
Блок RHS: Піррол одиниці II і III були об'єднані, щоб дати очікувану димерну одиницю з хорошою врожайністю. Ацилювання β-карбометоксипропіонілхлоридом дало блок RHS (12.17).
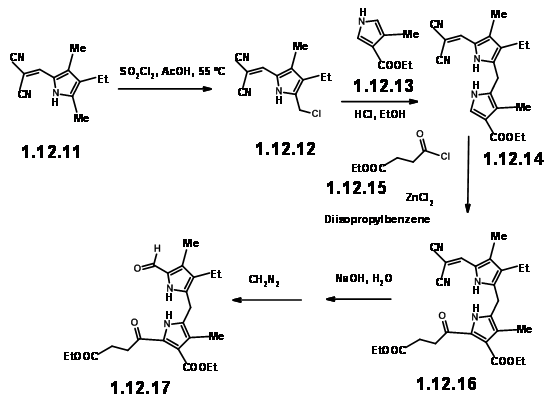
Маючи цей блок RHS (12.17) під рукою, вони спробували вирішальну циклізацію з легкодоступною моделлю LHS (12.18). При конденсації в кислотних умовах з подальшим окисленням йодом необхідний порфірин (12,19) міг бути отриманий з прийнятним виходом 25%.
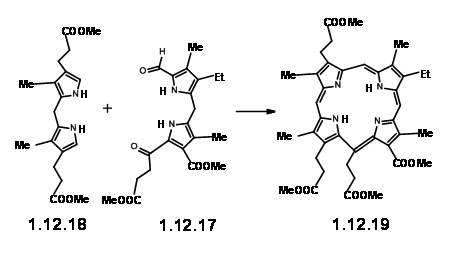
Заохочуючи цим результатом, вони пішли вперед, щоб синтезувати фактичну одиницю LHS (12,26).
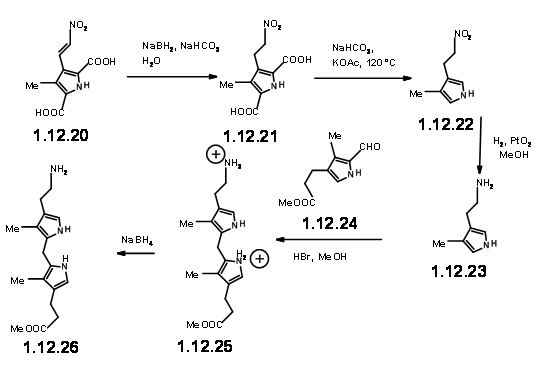
З готовим блоком LHS під рукою, вони подивилися на циклізацію цих двох одиниць. Вони зрозуміли, що ця конденсація може дати два продукти завдяки двом різним орієнтаціям реагентів. Хоча вихід цього продукту конденсації був порівнянний з попередньою конденсацією, вони побачили деякі недоліки.
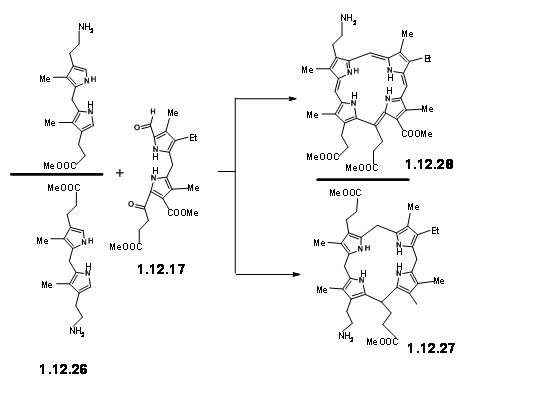
Урожайність була неприйнятною для їх прогнозованого призначення. Крім того, цей шлях призвів до утворення ізомерних продуктів, призначення структури яких створювало проблеми. Така «неелегантність» (вираз, який використовував RBW у своїй лекції) була неприйнятною для їхньої групи. Отже, вони вирішили заморозити рухливість двох одиниць, зв'язавши одиниці аміну та альдегіду в базу Шиффа (12.29).
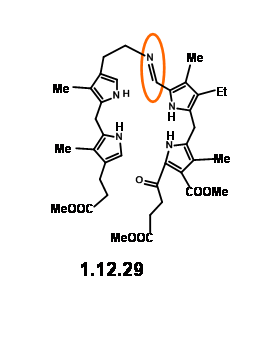
Однак це віртуозно визначене майстерність у молекулярній інженерії було нелегко досягти на практиці. Піррольні альдегіди, як правило, не були реактивними і можуть бути перетворені на основи Шиффа лише в умовах кислотного каталізу або буферизованих умов. Проблема виникла з блоку LHS, який виявився дуже чутливим до таких умов реакції. Ніяких слідів від очікуваного продукту конденсації не спостерігалося. Після кількох експериментів активація на альдегідній установці через основи Шиффа з подальшою конденсацією з блоком LHS через базовий обмін стала життєздатним підходом. Але відкриття, що такі бази Шиффа можуть бути

плавно перетворюється в тіоальдегід (12,30) забезпечується прорив. Цей тіоальдегід плавно конденсується з блоком LHS в нейтральних умовах з хорошим урожаєм. Ця «надзвичайно чутлива» основа Шиффа при кислотній обробці дала катіон (12.31), який був виділений як дибромід. Ця дикація була негайно окислена йодом до (12,32) і виділена у вигляді ацетаміду (12,33) при ацилюванні оцтовим ангідридом. Незважаючи на чутливість проміжних продуктів та швидкі подальші кроки, порфірин (12,33) може бути отриманий з хорошим урожаєм і може бути збільшений до декількох грам шкали.
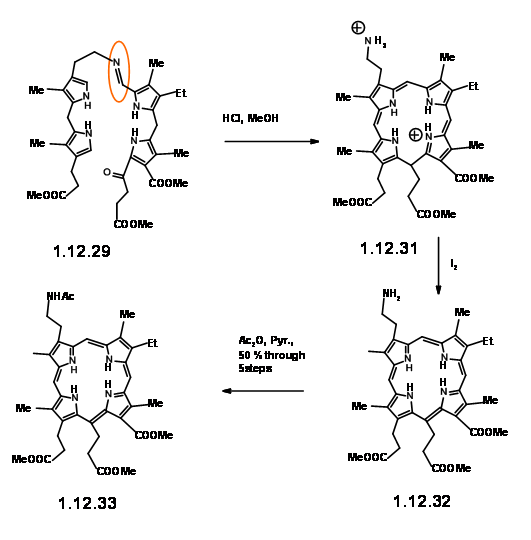
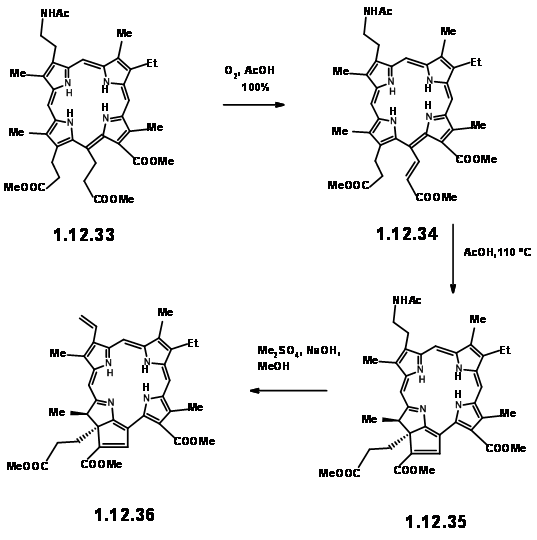
Коли порфірин (12,33) нагрівався в оцтовій кислоті в повітрі, відбувалася міграція першого водню, щоб дати (12,34). При нагріванні в оцтовій кислоті при 110 0С ланцюг при Cγ циклічно циклікували при С7, щоб дати відновлений кільцевий блок IV. Зверніть увагу, що орієнтація двох ланцюгів на кільці IV є транс-як очікувалося. На цьому етапі увага була переведена на вінілову групу на кільці I. Це було легко досягнуто шляхом вичерпної послідовності метилювання-елімінації Гофмана, показаної вище (12.36).
При впливі світла та повітря додаткове кільце легко розщеплюється, щоб дати піровиноградний ефір та формульну групу (12,37). Додатковий пірувіловий фрагмент був легко розщеплений в метанольних КОН, що призвело до метоксилактону (12.380. Сухий HCl дав гаміацеталь, який можна було б вирішити за допомогою методу хінінної солі, щоб дати (+) -Хлорин-5 (12,39). Діазометану етерифікації піддається альдегідної одиниці (12,40). Обробка формульної сполуки (12,40) HCN в триетиламіні дала ціанолактон (12,41), який при відновленні, етерифікації та гідролізі метанольної HCl дав (12,44), попередник продукту циклізації Дікмана.
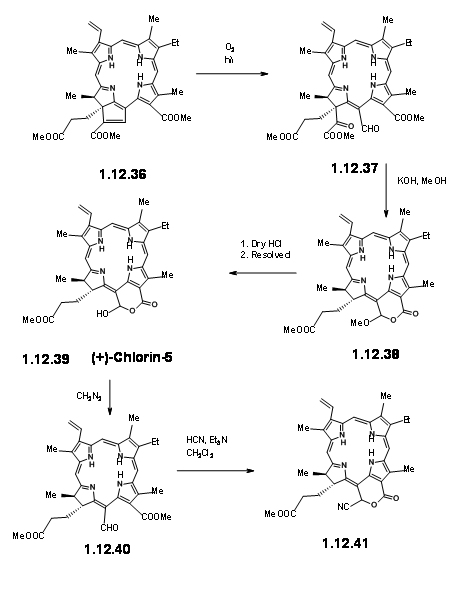
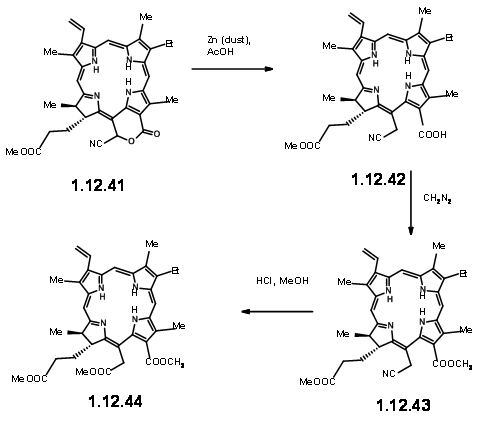
Звідси до (-) -Клорофіл α вже було відомо. А. Дікманн з подальшим введенням фітилової групи ферментатичними методами завершив синтез (Fortcher. Хім. Форш., 2, 538 (1952).
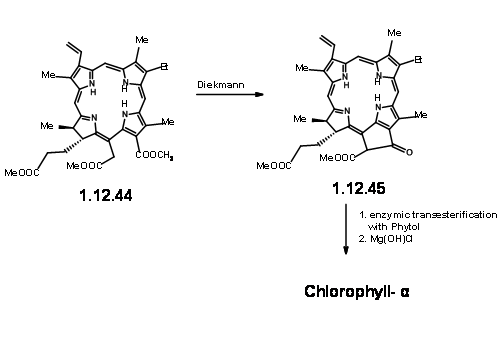
До цього дня цей шедевр в органічному синтезі має кілька особливостей, які утримують інтерес у любителів мистецтва і логіки в органічному синтезі.