4: Логіка синтезу
- Page ID
- 24294
«В органічному синтезі може бути АРТ» заявив неповторний монарх органічного синтезу професор Р.Б. Вудворд. Його школа відкрила кілька елегантних підходів, що охоплюють різноманітні складні структури і зламали нові підстави для визначення мистецтва органічного синтезу. «Якщо органічний синтез є галуззю науки, то яка логіка органічного синтезу?» дивувалися декільком іншим. Розробка концепції логічних підходів до синтезу розвивалася протягом останніх кількох десятиліть. Кілька сталвартів зосередили свою увагу на цій темі і намагалися розробити шаблон для визначення цієї логіки. Немає сумнівів, що всі ми, хто балується синтезом, вносять свій маленький шматочок у чудовому напрямку. Кілька імен виділяються в нашій свідомості своїми видатними внесками. Знатні внески надійшли від шкіл J.A.Marshal, E.J Wenkert, G. stock, S Hanessian, E.E. ван Тамален, С. Масамуне, Р.Б. Вудворд, E.J. Корі та кілька інших. Більш зосереджені на цій темі були внески від школи Е.Дж. Корі.
Період 1960 - 1990 став свідком еволюції цієї думки, і концепція розцвіла в повноцінну тему, яка зараз заслуговує окремого простору в навчальній програмі коледжу. Попередні розробки були зосереджені на ідеї АНТИТЕТИЧНИХ ПІДХОДІВ і удосконалювали мистецтво відключення через РЕТРОСИНТЕЗ. Це призвело до логічних підходів до побудови СИНТЕТИЧНИХ ДЕРЕВ, які узагальнювали різні можливі підходи до запропонованої цільової структури. Всі відключення можуть не привести до хороших маршрутів для синтезу. Після того, як синтетичне дерево було побудовано, окремі гілки були проаналізовані критично. Реакції були вивчені, щоб вивчити їх доцільність в лабораторії, їх механістичні шляхи були проаналізовані, щоб зрозуміти конформаційні та стереохімічні наслідки на результат кожного кроку, а також оцінювалися фактори часу/вартості запропонованих маршрутів. Можливі області підводних каменів були визначені, і література була критично відсканована, щоб переконатися, що розглянуті кроки вже відомі або здійсненні на основі відомої хімії. У деяких випадках модельні сполуки спочатку були побудовані для вивчення доцільності конкретної реакції, перш ніж приступати до синтезу складної молекулярної архітектури. Таким чином, довгий процес логічного планування зараз впроваджується до початку фактичного синтетичного проекту. Незважаючи на всі ці ретельні і тривалі приготування, досвідчений хімік все ще втомився від Дамоклів Меч синтезу а саме., ймовірний провал критичного кроку в запропонованому маршруті (ах), що призводить до повного провалу всього проекту. Всі досягнення - 10% натхнення і 90% потовиділення. Для цих відважних молекулярних інженерів, яких іноді ще називають хіміками, ці давно намальовані програми і можливі небезпеки невдач все ж варті, за потовиділення досить винагороди.
Знання механістичної органічної хімії, детальна інформація про мистецтво та науку функціональних групових перетворень, реакцій формування зв'язків та розщеплення, оволодіння методами поділу та очищення та обґрунтоване знання спектроскопічного аналізу є важливими основами синтезу. молекул. Синтетичний хімік також повинен знати про розвиток синтетичних стратегій, що генеруються роками для різних груп сполук, які включають в себе Правила та вказівки, що регулюють синтез. Оскільки органічна хімія має сильний вплив на розвиток інших сестринських дисциплін, таких як фармація, біохімія та матеріалознавство, здатність розуміти одну або кілька з цих областей та взаємодіяти з ними, використовуючи їх термінології, також є додатковою чеснотою для синтетичного хіміка. З досягненнями від синтезу напружених молекул (колись вважалися важкими (якщо не неможливими) для синтезу, до синтезу складних, високофункціоналізованих і нестабільних молекул, хімік-органік тепер міг би впевнено сказати, що він міг би синтезувати будь-яку молекулу, яка теоретично здійсненна. Це сучасний стан потужності органічного синтезу. Виходячи з завдання, покладеного на хіміка, він вибрав молекулу-мішень для дослідження і розробив відповідні шляхи синтезу.
Стратегії захисту та зняття захисту в органічних синтезах
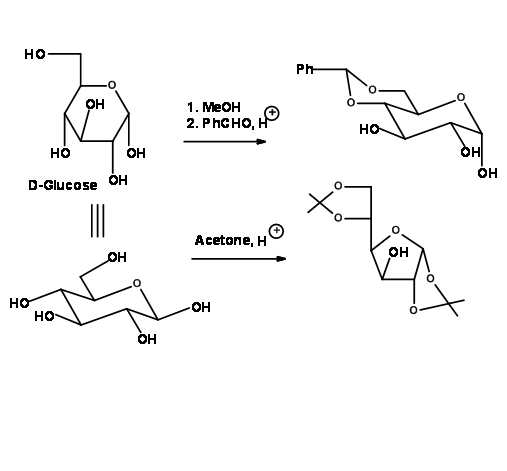
Для маніпулювання функціональними групами та формування нових ковалентних зв'язків ми використовуємо велику кількість Реагентів та Іменних Реакцій. У складних органічних синтезах вихідні матеріали і проміжні продукти в синтетичній схемі часто мають більше однієї реактивної функціональної групи. Кілька таких багатофункціональних будівельних блоків показані нижче, щоб проілюструвати цей момент (рис. 4.1.1). Під час роботи над таким комплексом
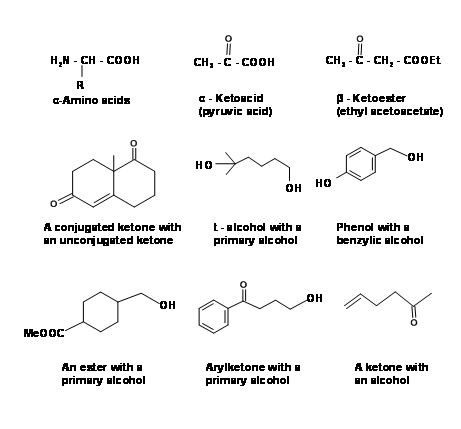
молекул, часто необхідно захистити деякі групи, щоб дозволити вибіркову роботу тільки в потрібних місцях. Органічні хіміки значною мірою покладалися на такі стратегії захисту/зняття захисту та старанно розробили протоколи захисту (маскування) та зняття захисту (зняття маскування). Ми б обговорили деякі важливі групи захисту в цьому розділі.
Перш ніж продовжити далі, тут слід підкреслити, що цей протокол слід застосовувати лише після критичного аналізу альтернативних варіантів. Це пов'язано з тим, що стратегія захисту/зняття захисту передбачає збільшення принаймні ще двох критичних кроків, додаючи до тривалості синтезу та, як наслідок, падіння загальної врожайності потрібної сполуки. У масштабних реакціях це призводить до величезного впливу на атомну економіку та витрати на забруднення синтетичного процесу. Все це виливається в збільшення загальної вартості кінцевої молекули препарату.
Стратегії захисту
Вибірковий реагент група/сайту: Захист/зняття захисту не завжди потрібно, коли ви бачите безліч функціональних груп. Ви можете вирішити проблему вибірковості за допомогою селективних реакцій/реагентів сайту. Вибравши відповідний селективний реагент відповідно до схеми на руках, ви могли вибірково атакувати тільки один з реактивних ділянок. Розглянемо олефіновий кетон (рис. 4.1.2). Зниження борогідриду натрію в метанолі як розчиннику може вибірково зменшити кето-групу до вторинного спирту
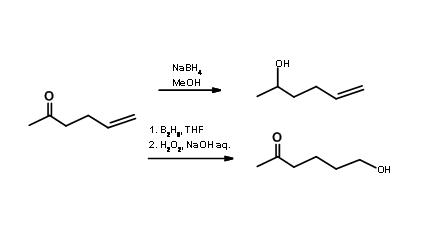
залишаючи олефін непорушеним. З іншого боку, диборановий реагент у THF як розчиннику був би реагентом вибору, коли бажано селективне відновлення на олефіновому фрагменті. Диборанове зниження олефіну в кілька разів швидше, ніж зменшення кетонів. Окислювальне розщеплення продукту бурану також є селективним. Таким чином, ви можете уникнути стратегії захисту/зняття захисту, використовуючи селективний реагент. У реакціях утворення зв'язків С - С ми стикаємося з декількома такими сайтоселективними реагентами. Одним з таких реагентів, широко використовуваних в дослідженнях, є реагент Віттіга. Вони атакують альдегід або кетон вибірково в присутності ефіру, нітрилу. олефіну і т.д..
Вибірковий захист
У випадку молекули, як 4.1.3A (рис. 4.1.3), що містить олефін та карбонову кислоту, група —COOH в кілька разів більш реактивна, ніж олефін до відновлення диборану.
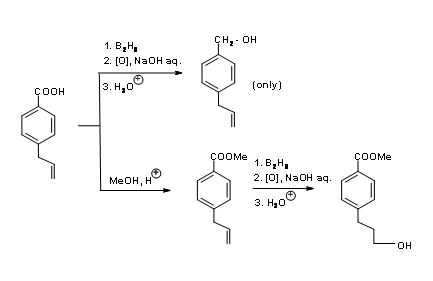
Гідроборація/окислення зменшує кислоту до первинного спирту, залишаючи олефін без впливу. З іншого боку, якщо вам потрібно селективне відновлення олефіну, кислотну групу потрібно обробляти за допомогою селективної послідовності захисту/зняття захисту, як показано на (рис. 4.1.3)
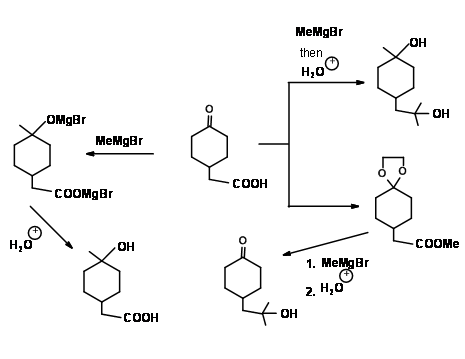
З'єднання 4.1.4A ілюструє кілька важливих моментів у протоколі захисту/зняття захисту. Обидві функціональні групи могли реагувати з реагентом Гріньяра. Група карбонових кислот спочатку реагує з одним молем реагенту Гріньяра, щоб дати карбоксилатну аніонну сіль. Цей аніон більше не реагує з реагентом. Коли в реакційну суміш додають два молі реагенту Гриньяра, другий моль атакує кетон, щоб дати третинний спирт. На водній обробці кислотна група регенерується. Таким чином, перший моль реагенту забезпечує селективний перехідний захист для групи —COOH. Після етерифікації кислотної групи така селективність по відношенню до цього реагенту втрачається. Реагент атакує на обох ділянках. Якщо реакція бажана тільки на місці ефіру, кето-група повинна бути вибірково захищена як ацеталь. На наступному етапі проводиться реакція гриньяра. Зараз реагент має тільки одну групу, доступну для реакції. При обробці кислотою захист металу в проміжному з'єднанні також гідролізується до регенерації кето- групи.
Ортогональний захист або диференціальний захист
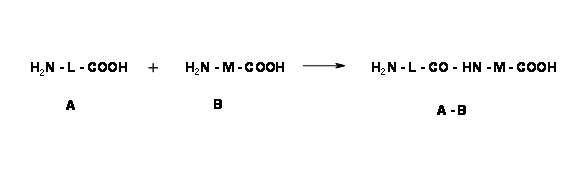
Ортогональний захист - це стратегія, яка дозволяє знімати захист декількох захисних груп по одній, кожна з виділеним набором реагентів і/умов реакції, не впливаючи на іншу. Ця методика найкраще ілюструється утворенням пептидних зв'язків та пов'язаними з ними реакціями депротекції. Амінокислота має дві функціональні групи —N H 2 і —COOH. Коли дві амінокислоти (А і В) реагують в умовах реакції конденсації пептидного зв'язку, суміш дипептидів 4 (принаймні) може утворитися, як показано нижче.
\[ \ce{ A + B \rightarrow A-A + A-B + B-A + B-B}\]
Якщо нас цікавить лише один продукт A — B, ми повинні робити вибірковий захист та селективну дезахист у належній послідовності. Розглянемо наступну реакцію формування пептидних зв'язків.
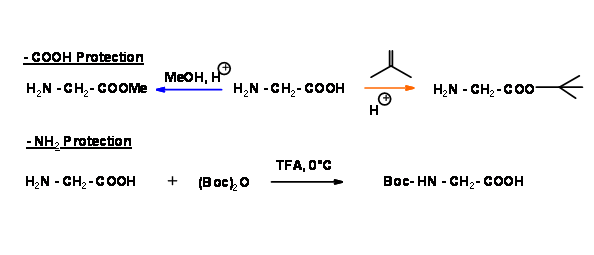
Для того, щоб отримати тільки один продукт A — B, ми повинні захистити N — термінал 'A' і C — термінал 'B'. Давайте уважно розглянемо дві різні схеми формування дипептидів. У наступній послідовності C — термінал захищений двома різними способами для однієї амінокислоти. Для другої амінокислоти N — термінал захищений кислотною лабільною Boc- захистом.
На наступному етапі дві монозахищені амінокислоти з'єднуються, як показано нижче.
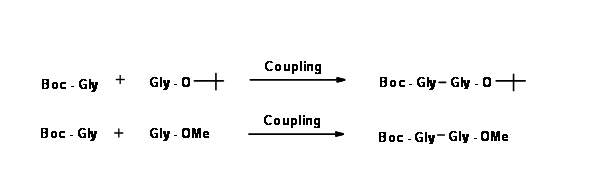
Уважно придивіться до обох продуктів. У першому продукті обидва захисту чутливі до кислоти. Якщо кінцевий продукт бажаний - це беззахисний дипептид, це дійсно короткий шлях.
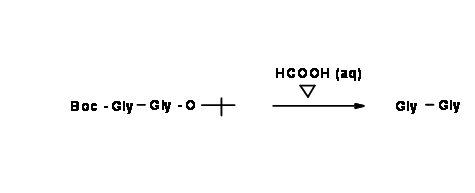
Якщо бажаний продукт є монозахищеним дипептидом, то переважною реакцією є селективна депротекція. Це можливо лише тоді, коли ми використовуємо вихідні сполуки, які диференційно захищені. Це називається ортогональним захистом.
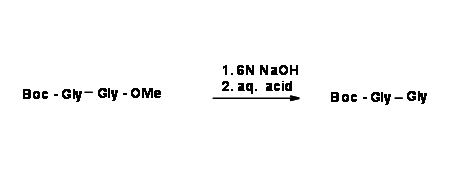
Подібні методи доступні і для інших функціональних груп, а також. Давайте тепер дізнаємося більше про захист/зняття захисту для деяких важливих функціональних груп.
Захист R — Група COOH
У вступі ми бачили, що карбоксилатний іон забезпечує захист від атаки реагентів Гріньяра на цей карбонільний вуглець. Однак цього недостатньо для великої кількості різноманітних реагентів. 2-оксазоліни Мейєра маскують кислотну функцію, активуючи α- положення для реакції літіювання. Використання цієї групи в якості захисту для групи —COOH зустрічається рідко.
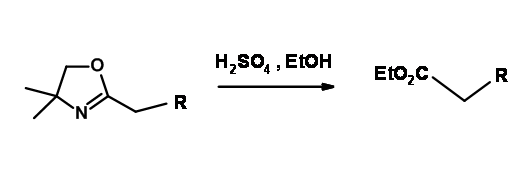
Захист альдегідів і кетонів
Оскільки спирти, альдегіди та кетони є найбільш часто маніпульованими функціональними групами в органічному синтезі, велика робота з'явилася в їх стратегіях захисту/зняття захисту. У цій дискусії давайте зосередимось на класах груп захисту, а не на вичерпному поводженні з усіма захистами.
Ацетали
Існує два загальних методу введення даного захисту. Транскеталяція - метод вибору, коли бажані ацетали (метали) з метанолом. Ацетон є побічним продуктом, який доводиться видаляти, щоб змістити рівновагу в праву сторону. Це досягається шляхом рефлюксу з великим надлишком ацетонідного реагенту. Утворився ацетон постійно переганяють. У разі циклічних діолов утворилася вода безперервно видаляється за допомогою конденсатора Декана-Лелеки (рис. 4.1.6).
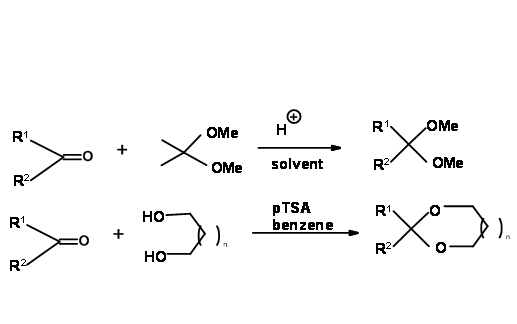
Швидкість утворення кеталів з кетонів і 1,2-етандіолу (етиленгліколю), 1,3-пропандіолу і 2,2-диметил-1,3-пропандіолу різні. Так само і реакція дететаляції. Це дозволило хімікам вибірково працювати в одному центрі. Наступні приклади з стероїдної хімії ілюструють ці моменти (Рис. 4.1.7).
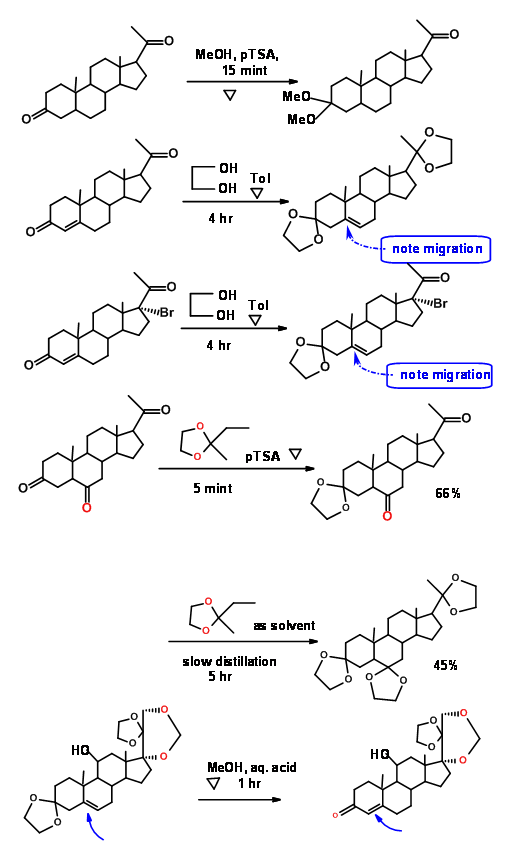
Попит на процеси зеленої хімії спонукав пошук нових зелених процедур. Деякі приклади з недавньої літератури наведені тут (рис. 4.1.8).
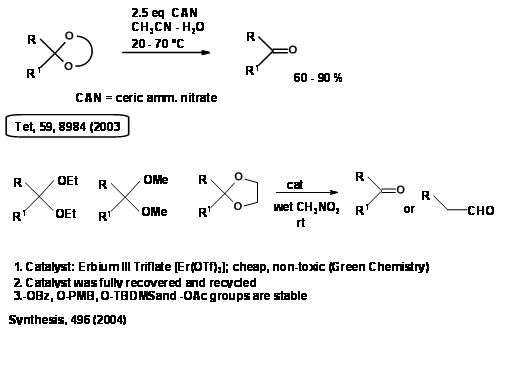
Тіокетали
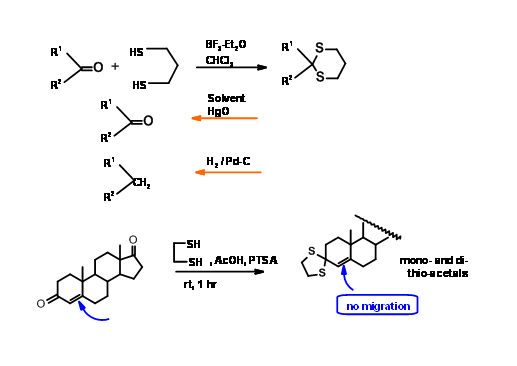
У порівнянні зі своїми кисневими аналогами тіокетали помітно відрізняються за своєю хімією. Утворення, а також зняття захисту сприяють відповідні кислоти Льюїса. Тіоацетали помітно стабільні в умовах дететаляції, тим самим відкриваючи шлях для селективних операцій у двох різних центрах. При залученні кон'югованих кетонів утворення металів (а також депротекція) протікає з міграцією подвійних зв'язків. З іншого боку, тіокетали утворюються і дететаліруются без подвійної міграції зв'язку (рис. 4.1.9).
Захист аміногруп (-NH2 & —NH)
N-ацетил (N - COC H 3), N - бензоїл (N - CoPH) Захист
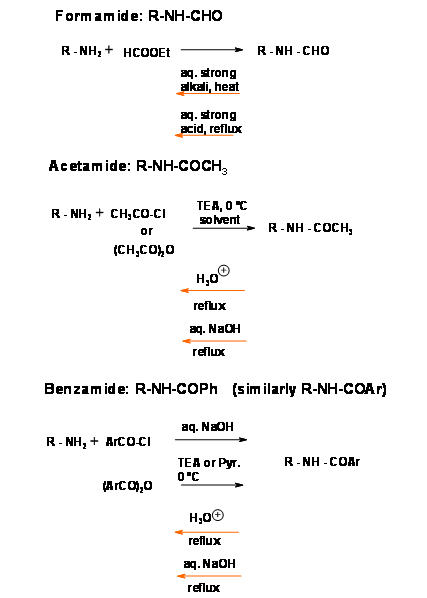
Це класичні захисні групи для первинних і вторинних амінів. Реагенти дешеві, а протокол простий. Такі аміди, як правило, потребують кардинальних умов для зняття захисту, хоча врожайність, як правило, хороша (рис. 4.1.10). Стандартною процедурою є рефлюкс у водній лузі або водній мінеральній кислоті. Через різкі умови, в цій процедурі слід дотримуватися обережності, щоб уникнути рацемізації. Аміди - це, як правило, кристалічні тверді речовини, які легко очищаються кристалізацією. Коли захист впроваджується на ранніх стадіях довгої синтетичної схеми і бажаний дуже стабільний захист (як при синтезі нуклеотидів), амід є найбільш кращим захистом.
Досліджено ще кілька лабільних амідних зв'язків. Особливий інтерес представляють аміди трифтороцтової кислоти. Введення, а також розщеплення є простим і м'яким {Рис. 4.1.11).
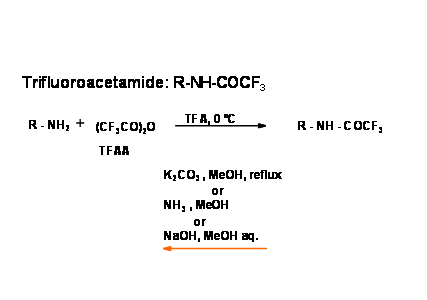
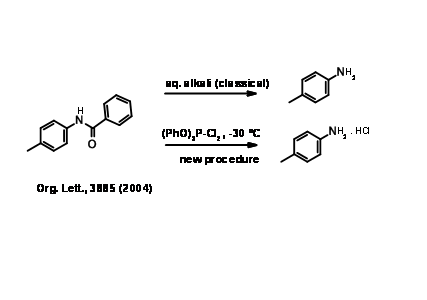
Нещодавній звіт про гідроліз амідів наведено нижче.
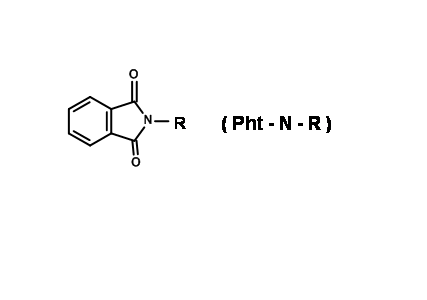
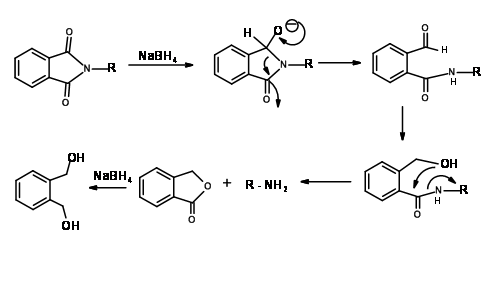
N - Захист від фталоїлу (N - Pht)
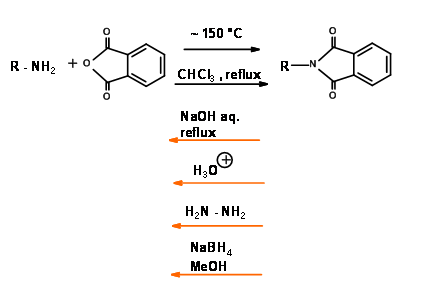
Механізм для NaB H 4 Зменшення N — Pht
N - складні ефіри карбонової кислоти як захисні групи
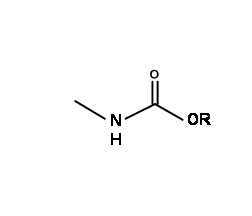
Як описано вище, амідні зв'язки дуже міцні. З іншого боку, ефірні зв'язки легко розщеплюються м'якими базовими умовами. Карбоетокси-захист на аміні має амідний зв'язок, а також ефірний зв'язок. Оскільки N — COOH групи, отримані при гідролізі, дуже нестабільні, такий захист забезпечує велике сімейство захисних груп для первинних і вторинних амінів.
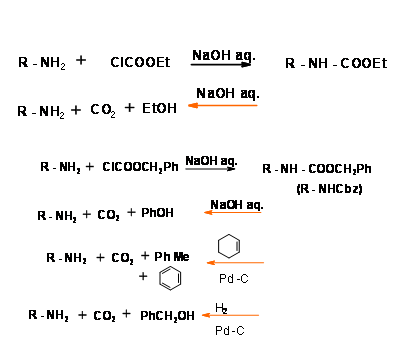
N - Карбоетоксикарбон (N - CooEt) та карбобензилоксикарбоніл (N - CooCh2pH) (N - Cbz або N - Z) Захист:
Ці групи легко вводяться за допомогою відповідних ефірів хлороформатів. Ангідриди або змішані ангідриди в м'яких основних умовах. Обидва ці захисту можна було зняти при тривалому перемішуванні з основою при кімнатній температурі. Хоча м'який, деяка рацемізація іноді спостерігається. Захист N - Cbz має додаткову перевагу в тому, що він може бути легко розщеплений в умовах гідрогенолізу (рис. 4.1.14). N - Cbz Захист, однак, стійкий до кислих умов. Порівняйте це з захистом —Boc, розглянутим нижче.
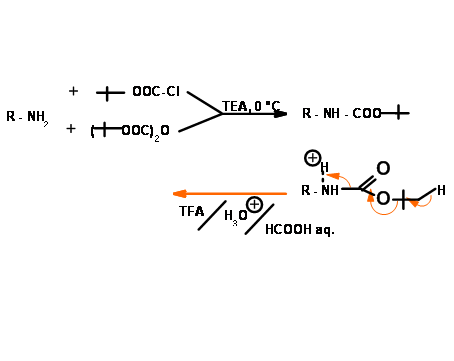
Трет-бутилоксикарбонільний захист (N - CoOBut, N - Boc)
Трет-бутилоксикарбонільний захист може бути введений і видалений в дуже м'яких умовах кислоти. Цей захист стійка до лугу і гідрогенолізу (рис. 4.1.15). Таким чином, N — Z і N — Boc є компліментарними як захисні групи.
N - Фторметиленоксикарбонільний захист (Fmoc)
Ця ультрафіолетова активна захисна група дуже популярна в протоколах твердофазного синтезу пептидів (SPPS). Захист, а також заходи зняття захисту протікають в м'яких умовах при хорошій врожайності (рис. 4.1.16).
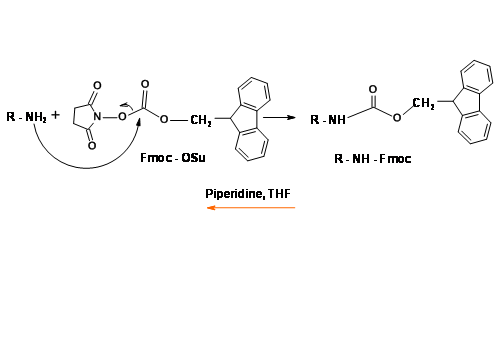
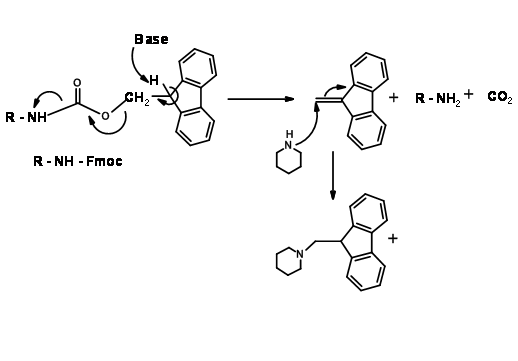
Механізм зняття захисту Fmoc показаний на (рис. 4.1.17)
N - Силізація
Силілювання є загальним захистом активного водню на гетероатомах. У випадку зв'язку N - Si фториди четвертинного амонію розщеплюють цей зв'язок (рис. 4.1.18).
N - Тостиляція (N — Tos)
Такий захист дуже стійка. N - Тозилювання легко здійснюється за допомогою процедури хлориду кислоти. Він розщеплюється реакцією розщеплення сольватів електронів. Коли ця група приєднується до первинного аміну, група —NH стає дуже кислою (рис. 4.1.19).
Захист — OH Групи
Ацетати (— Ac) і бензоати (— Obz)):
Хімія захисту групи OH була широко досліджена. Класичною захистом є утворення ефірів аліфатичної і ароматичної карбонових кислот. Ароматичні ефіри порівняно важко гідролізувати при м'якому базовому стані. Це дає можливість для вибіркових протоколів зняття захисту (рис. 4.1.20). Зверніть увагу, що цей захист чутливий до кислотних, а також базових умов.
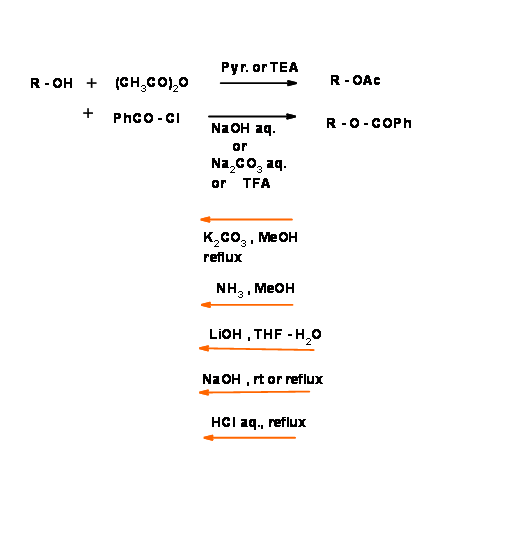
Метиловий (- ОЕ) і бензиловий (R - On) ефіри
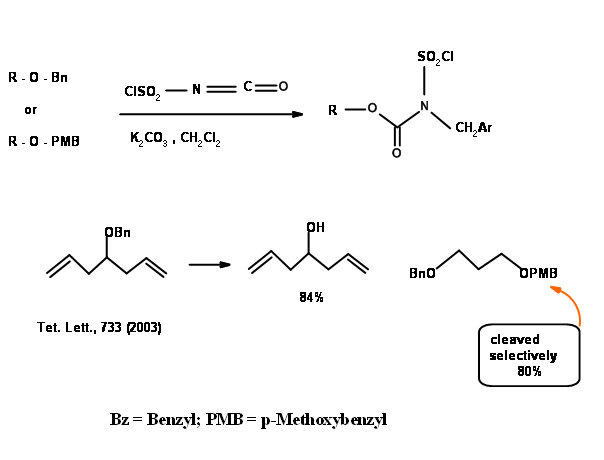
Ефірна група є однією з найбільш стабільних функціональних груп. Отже, ця група була найбільш прихильною захисною групою. Зняття захисту було проблемою. На початку ХХ століття єдиною процедурою було рефлюксування водним HI або HBr. В останні роки з'явилося кілька нових процедур для ефективного видалення в м'яких умовах. Особливістю бензилових ефірів є те, що цей захист легко знімається в умовах нейтрального гідрогенолізу (рис. 4.1.21). Замінники, такі як - oME або - NO2, можуть бути введені на бензольне кільце, щоб змінити реакційну здатність на місці захисту.
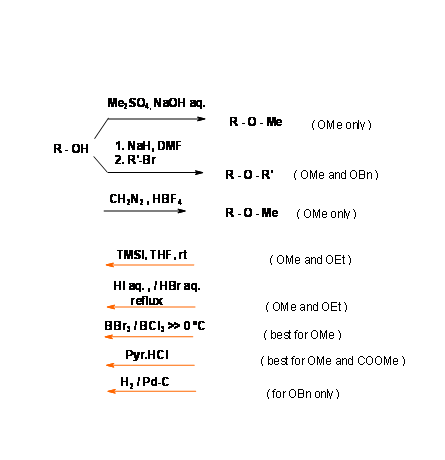
Коли олефін може конкурувати в процедурі гідрогенолізу, наступна послідовність, здається, є альтернативною процедурою (рис. 4.1.22).
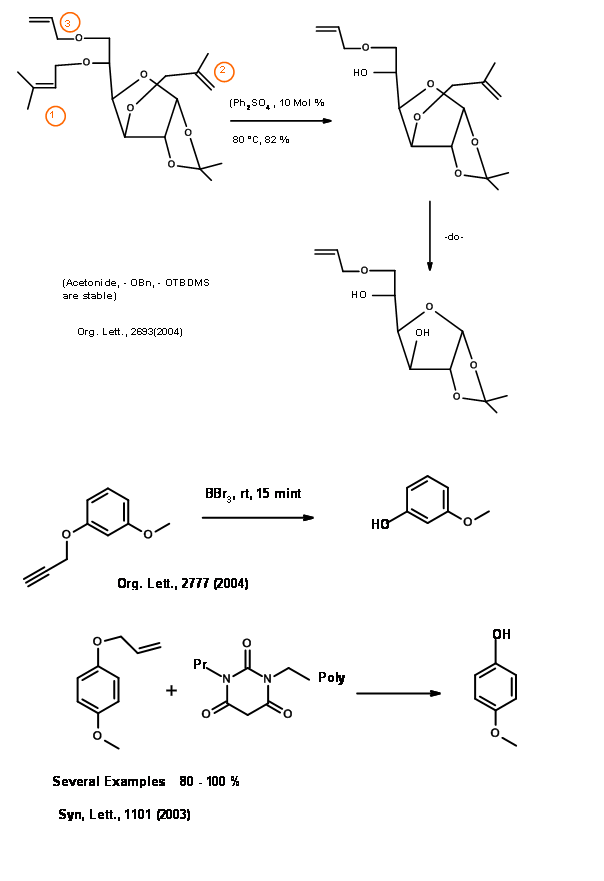
Алліловий ефір - це недавнє введення в захист OH. Універсальність цього захисту можна побачити на наступних прикладах (рис. 4.1.23).
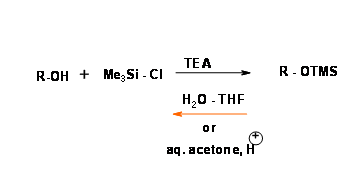
Силілові ефіри (R — OSi R 3)
Киснево-кремнієвий сигма-зв'язок стійкий до реагентів літію та Гріньяра, нуклеофілів та гідридних реагентів, але дуже нестійкий до води та м'яких водних кислот та базових умов. Силіловий ефір вторинного спирту менш реактивний, ніж первинний спирт. O — триметилсиліл (O — SiME3) був першим захистом цього класу. (Рис. 4.1.24).
Заміна метильної групи іншими алкіловими і арильними групами дає велику різноманітність силілового ефіру з різним ступенем стійкості до гідролізу (рис. 4.1.25).

Наступні приклади ілюструють селективність при утворенні і гідролізі цієї групи (рис. 4.1.26).
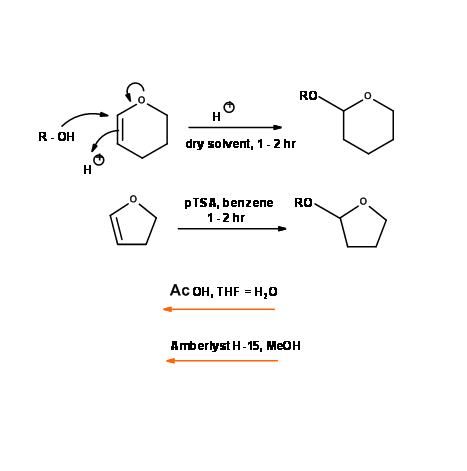
Тетрагідропіраніловий ефір (— OTHP) та тетрагідрофураніловий ефір (— OTHF)
Ці захисні групи для спиртів насправді є ацетальними. Вони синтезуються за допомогою дигідропірану (ДГП) і дигідрофурана (ДГФ) відповідно. Вони поводяться як ацетали за своєю стабільністю і розщепленням (рис. 4.1.27). Швидкість утворення і розщеплення для цих двох груп відрізняються, що знаходить застосування для диференціального захисту спиртів.
Ці захисні групи знайшли широке застосування в синтезі. Однак незабаром спостерігалися два основних недоліки.
- Під час введення цього захисту генерується нова стереоточка. Хоча це не актуально з точки зору молекули-мішені, в хіральних молекулах це створило діастереомерні проблеми в спектроскопії (ЯМР та МС) та хроматографії.
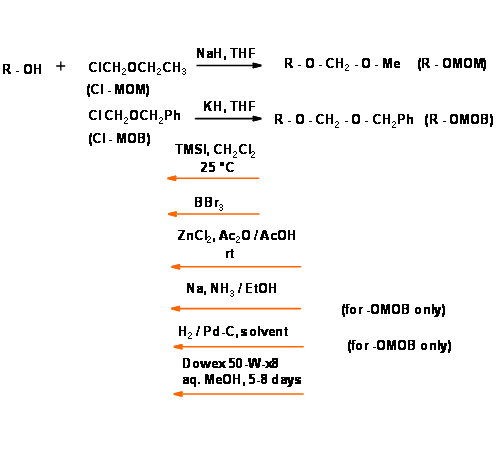
- Ці ефіри іноді спричиняли вибухи в процедурах гідробірації через утворення перекису. Проблема діастереомерів була вирішена введенням О-метиленоксиметилового ефіру (— O — МОМ) та O — метиленеоксибензилового ефіру (R — O — MOB) (рис. 4.1.28). Зараз доступні кілька інших модифікацій.
Захист від віка — Діоли
При реакції з бензальдегідом або ацетоном з відповідним кислотним каталізатором вік-діоли утворюють циклічні ацетали. Це насправді є доказом існування вік-діолів в молекулі. Вони є ацетальними захистами і тому поводяться як ацетали у своїй хімії (рис. 4.1.29)
Висновок
Наведені вище обговорення - це лише проблиск великої літератури на цю тему. Коли в молекулі присутні більше однієї конкуруючих функціональних груп, може знадобитися ввести в синтетичній схемі хоча б одну ступінь захисту і один етап зняття захисту. Це додає не тільки довжини синтетичної схеми, але і до вартості кінцевого з'єднання. Зі зростанням обізнаності в зеленій хімії хіміки намагаються звести цей протокол до мінімуму або бажано уникнути цього взагалі. У літературі відомо кілька захисних беззахисних синтезів натуральних продуктів. Ми б обговорили цю тему в кінці цього розділу.
Подальше читання
- Грін Т. В., Захисні групи в органічному синтезі. Вілі. Н. У., (1980), (1991).
- Сміт М.Б., Органічний синтез, Макгроу-Хілл Inc, Н.Ю., (1994).
- Djerassi C., Стероїдні реакції —Контур для органічних хіміків, Holden-Day nc. Сан-Франциско) 1963).
- Розширена органічна хімія: принципи, інструменти та логіка синтезу, Р.Баладжі Рао, Vishal Publishing Co., Джаландхар, Індія (2012).
- Амінокислоти, пептиди та білки в органічній хімії, Том 4, Ред Ендрю Хьюз (2011) Wiley-VCH; Реакції захисту, В.В.
4.2 Відключення облігацій
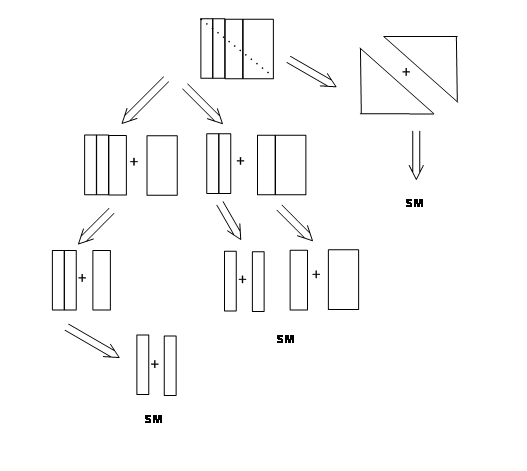
Вибравши молекулу TARGET для синтезу, наступною вправою є складання синтетичних планів, які б узагальнили всі розумні шляхи її синтезу. Протягом останніх кількох десятиліть хіміки працювали над процесом під назвою РЕТРОСИНТЕЗ. Ретросинтез можна охарактеризувати як логічне відключення стратегічних зв'язків таким чином, що процес поступово призведе до легкодоступного вихідного матеріалу (ів) через кілька синтетичних планів. Кожен план таким чином еволюціонував, описує «МАРШРУТ», заснований на ретросинтезі. Кожне відключення призводить до спрощеної конструкції. Логіка таких роз'єднань є основою для ретроаналізу заданої молекули-мішені. Натуральні продукти забезпечили хіміків великою різноманітністю структур, що володіють складними функціональними можливостями і стереохімією. Цей напрямок забезпечив низку складних завдань для розробки цих концепцій. Принцип підкреслення при розробці логічних підходів для синтетичних маршрутів дуже схожий на наступну просту задачу. Давайте розглянемо наступний великий блок, який виготовляється шляхом складання декількох невеликих блоків (рис. 4.2.1). Ви можете легко побачити, що великий блок може бути розбитий різними способами, а потім зібраний, щоб дати той же оригінальний блок.
Тепер спробуємо розширити той же підхід для синтезу простої молекули. Давайте розглянемо три можливі «відключення» для циклогексанового кільця, як показано на малюнку 4.2.2.
У наведеному вище аналізі ми спробували розробити три способи відключення шестичленного кільця. Чи створили ми таким чином три шляхи синтезу циклогексанового кільця? Чи мають такі відключення хімічний сенс? Передумови органічного хіміка повинні дати йому можливість зчитувати процес як хімічну реакцію в зворотному (або «ретро-») напрямку. Точки у вищезазначених структурах можуть представляти іон карбонію, карбоніон, вільний радикал або більш складну реакцію (наприклад, перициклічну реакцію або перестановку). Застосування такого хімічного мислення може відкрити кілька правдоподібних реакцій. Давайте подивимося на шлях b, який став результатом розщеплення однієї сигма-зв'язку. Один тільки аніонний маршрут циклізації виставляє кілька кандидатів як відповідні проміжні продукти для формування цього зв'язку. Наведений вище аналіз описує лише три шляхи з великої кількості альтернативних шляхів розщеплення, які доступні. Розширений аналіз, показаний нижче, вказує на більше таких можливостей (рис. 4.2.3). Кожен такий проміжний продукт може бути підданий подальшому процесу відключення, і процес тривав, поки ми не досягнемо досить невеликих, легко доступних вихідних матеріалів. Таким чином, може бути побудовано повне «СИНТЕТИЧНЕ ДЕРЕВО», яке б узагальнило всі можливі шляхи для даної молекули мішені.
4.3 Ефективність маршруту
Маршрут вважається ефективним, коли «загальний вихід» загального процесу є найкращим серед усіх досліджуваних маршрутів. Це буде залежати не тільки від кількості кроків, задіяних у синтезі, але і від типу стратегії, яку дотримується. Стратегія може включати в себе «лінійний синтез», що включає лише послідовні кроки або «конвергентний синтез», що включає менше послідовних кроків. Малюнок 4.3.1, показаний нижче, зображує кілька візерунків, які можна було б розпізнати в таких синтетичних деревах. Коли кожен процес відключення призводить лише до одного можливого проміжного, і процес протікає таким чином.
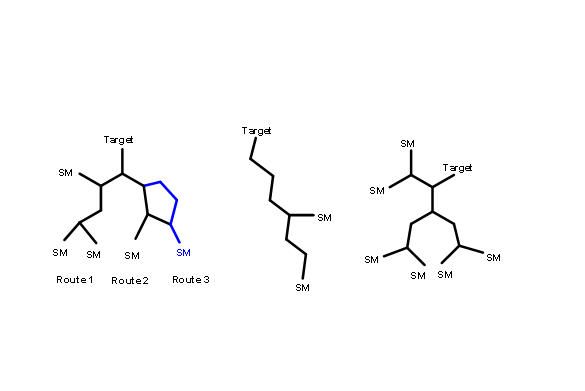
аж до одного набору вихідних матеріалів (СМ) процес називається лінійним синтезом. З іншого боку, коли проміжний може бути відключений двома або більше способами, що ведуть до різних проміжних продуктів, в плані відбувається розгалуження. Процеси можна було б продовжувати аж до SMS. У таких маршрутах різні гілки синтетичних шляхів сходяться в бік проміжного. Такі схеми називаються збіжними синтезами.
Блок-схеми, наведені нижче (рис. 4.3.2), зображують гіпотетичний 5-ступінчастий синтез за двома вищезазначеними стратегіями. Припускаючи дуже хороший вихід (90%) на кожному кроці (це рідко спостерігається в реальних проектах), синтез лінії дає 59% загальний вихід, тоді як конвергентний синтез дає 73% загального прибутку за ту ж кількість кроків..
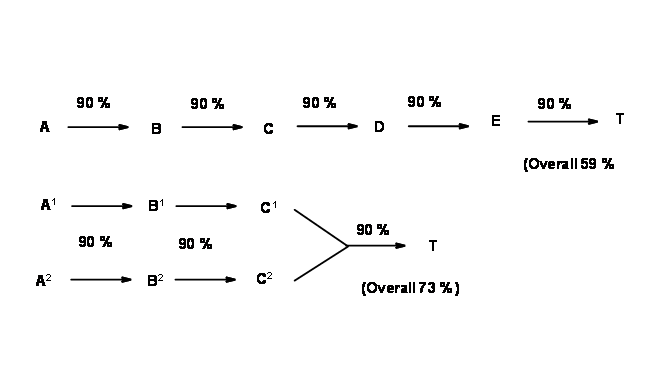
4.4 Проблема замісників і стереоізомерів
Ситуація ускладнюється, якщо розглядати можливість утворення небажаних ізомерів, що утворюються на різних етапах синтезу. Загальний вихід значно знижується для синтезу правого ізомеру. Тому переважними є реакції, які дають поодинокі ізомери (діастереоспецифічні реакції) у хороших врожаях. Деякі реакції, такі як реакція Дільса Вільхи генерують кілька стереоточок (точок, в яких генеруються стереоізомери) одночасно в одному кроці в дуже передбачуваному порядку. Такі реакції високо цінуються при плануванні синтетичних стратегій, оскільки за один крок вводяться кілька бажаних структурних особливостей. Там, де один чистий енантіомер є мішенню, ситуація знову складна. Чиста сполука на заключному етапі все ще може мати 50% небажаного енантіомера, що призводить до різкого падіння ефективності маршруту. У таких випадках бажано відокремлювати оптичні ізомери якомога раніше, по синтетичному маршруту. Це головна заслуга підходу Хірона, в якому правильний вихідний матеріал вибирається з легко доступного, дешевого «хірального басейну». Ми б обговорили цей аспект після того, як зрозуміли логіку синтезу планування. Враховуючи ці параметри, тепер ви можете визначитися з найбільш ефективним маршрутом для будь-якої заданої мети.
Молекули, що цікавлять, часто є більш складними, ніж звичайне циклогексанове кільце, розглянуте вище. Вони можуть мати замісники і функціональні групи в заданих точках і навіть конкретних стереохімічних точках. Конструкція з синтетичного дерева повинна ідеально вмістити всі ці параметри, щоб дати ефективні траси. Давайте розглянемо трохи більш складний приклад, показаний на малюнку 4.4.1. Кетон 4.4.1A необхідний як проміжний продукт у синтезі. На відміну від звичайного циклогексану, розглянутого вище, схема заміщення і кето- група в цій молекулі накладають деякі обмеження на процеси відключення.
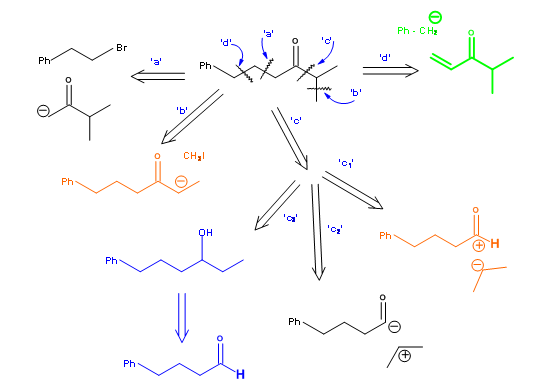
Розщеплення a: Цей шлях передбачає напад аніону метилізопропілкетону на бром-компонент. Розщеплення b: Цей шлях передбачає просте регіональне метилювання більшого кетону, який несе всі інші структурні елементи. Розщеплення c: Цей маршрут передбачає три різні можливості. Маршрут С-1 передбачає блок ациклонію, який може надходити з галогеніду кислоти або ефіру. Маршрут С-2 має на увазі реакцію ампулунга на ацильної одиниці. Маршрут С-3 передбачає окислення вторинного спирту, яке можна було отримати за допомогою реакції типу Гриньярда. Розщеплення d: Це означає доповнення Майкла.
Кожен з цих маршрутів може бути додатково розроблений назад, щоб завершити синтетичне дерево. Це лише кілька правдоподібних маршрутів, щоб проілюструвати важливий момент, що деталі структури обмежують можливі розщеплення деякими стратегічними точками. Значний внесок у планування органічних синтезів надійшов від школи Е.Дж. Корі. Ці розробки були зібрані Корі в книзі під назвою ЛОГІКА ХІМІЧНОГО СИНТЕЗУ. Ці та кілька супутніх презентацій на цю тему слід сприймати як орієнтири. Вони розроблені після аналізу більшості відомих підходів, опублікованих в літературі, і виявлення закономірності в логіці. Їм не потрібно обмежувати простір для нових можливостей. Деякі з важливих стратегій викладені нижче.
4.5 Попереднє сканування
Коли синтетичний хімік дивиться на дану Мішень, він повинен спочатку обміркувати деякі попередні кроки, щоб спростити проблему на руках. Чи є молекула полімерною? Подивіться, чи може вся молекула бути розділена на мономерні одиниці, які можуть бути з'єднані відомою реакцією. Це легко помітити у випадку з пептидами, нуклеотидами та органічними полімерами. Це також може бути вірно для інших натуральних продуктів. У таких молекулах, як C-токсиферин 1 (4.5.1A) (рис. 4.5.1), точка димеризації очевидна. У ряді інших випадків для ідентифікації мономерних одиниць потрібно більш глибоке розуміння, як у випадку з Уснінової кислотою (4.5.1B). У випадку з макролідним антибіотиком Нонактин (4.5.1C) ця стратегія знижує можливості синтезу мономерної одиниці (4.5.1D). Загальна структура має симетрію S4 і є ахіральною, хоча зібрана з хиральних попередників. Обидві (+) -неактична кислота і (−) -неактична кислота (4.5.1D) необхідні для побудови макроциклу, і вони з'єднуються головою до хвоста в змінному (+) - (−) - (+) - (−) візерунок. (див. J. Am. Хім. Соц., 131, 17155 (2009) та посилання, наведені в них).

Чи вже вирішена частина структури? Критичне вивчення літератури часто може виявити, що вирішена одна і та ж молекула або тісно пов'язана. Р.Б. Вудворд синтезував (4.5.2C) як ключовий проміжний продукт в елегантному синтезі резерпіну (4.5.2A). Це ж проміжне з'єднання (4.5.2C) стало ключовим вихідним з'єднанням для Velluz та ін., в синтезі дезерпідину (4.5.2B) (рис. 4.5.2).
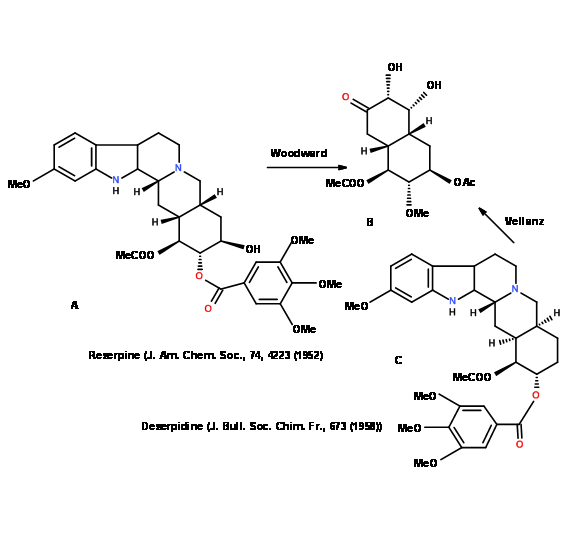
Такі стратегії скорочують час, необхідний для синтезу нових кандидатів на наркотики. Ці стратегії часто використовуються в хімії натуральних продуктів і хімії ліків. Після завершення попереднього сканування молекула-мішень може бути від'єднана в Стратегічних облігаціях.
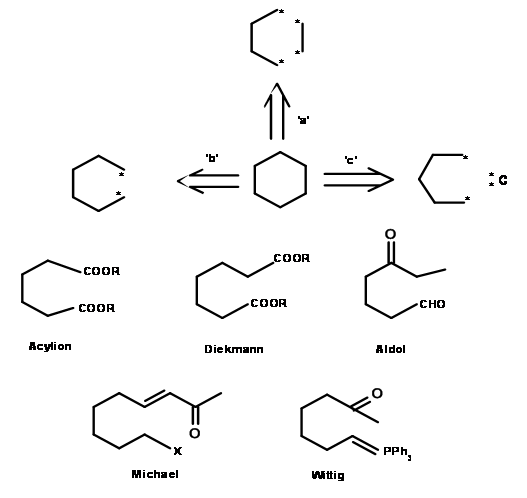
4.6 Стратегічні облігації, ретрони та перетворення
СТРАТЕГІЧНІ ОБЛІГАЦІЇ - це облігації, які розщеплюються, щоб прийти до відповідних вихідних матеріалів (SM) або SYNTHONS. З метою відключення зв'язку Корі припустив, що структуру можна класифікувати відповідно до підструктур, породжених відомими хімічними реакціями. Він назвав підструктури RETRONS, а хімічні перетворення, які генерують ці Ретрони, називалися ТРАНСФОРМАМИ. Короткий список перетворень і Ретронів наведено нижче (ТАБЛИЦЯ 4.6.1). Зверніть увагу, що коли перетворення генерують Retrons, продукт може мати нові STEREOPOINTS (стереохімічні деталі) генеруються, які можуть потребувати критичної оцінки.
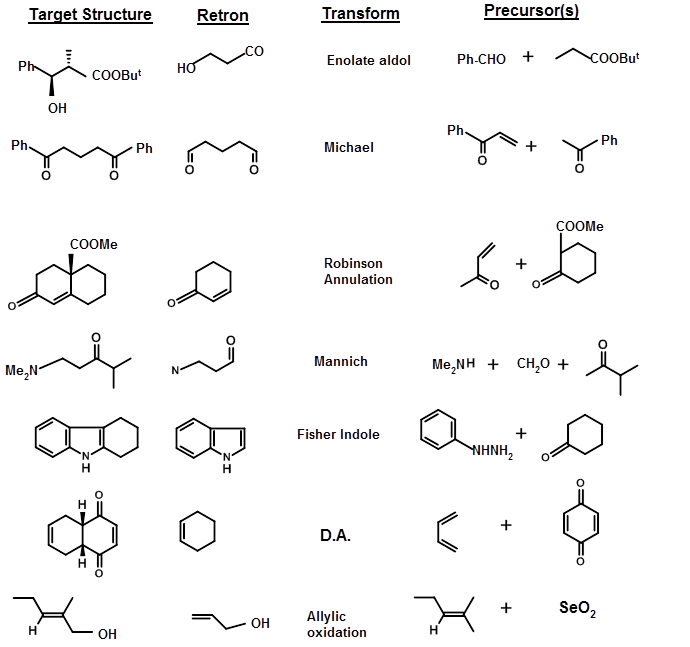
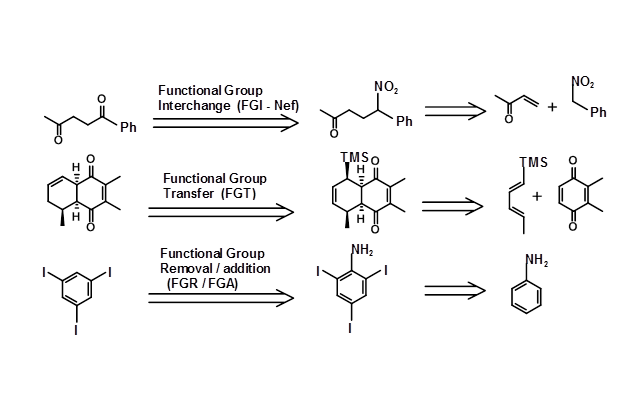
Структура цілі може бути такою, що Ретрон та відповідні перетворення можуть бути легко візуалізовані та безпосередньо застосовані. У деяких випадках перетворення або Ретрони можуть бути не очевидними. У декількох синтезах перетворення не спрощують молекулу, але полегшують процес синтезу. Наприклад, кето-група може бути сформована шляхом модифікації блоку -CH-N O 2 за допомогою реакції Nef. Це генерує новий набір пар Retron/transforms. Кілька таких перетворень перераховані нижче разом з номенклатурою, запропонованою Корі (рис. 4.6.2).
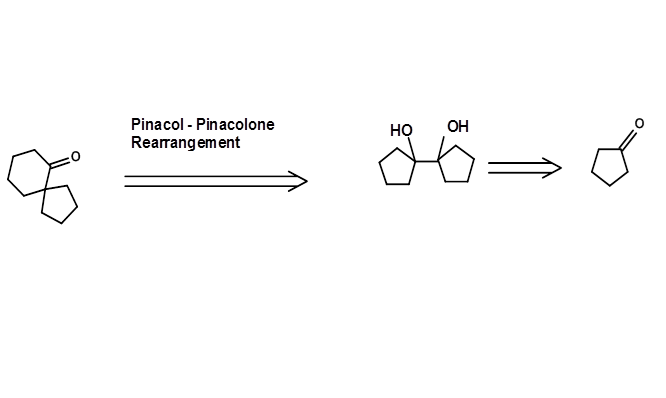
Реакція перестановки може бути потужним методом для створення відповідних нових підструктур. У наступному прикладі відповідний Пінакол-ретрон, необхідний для перестановки, отримують за допомогою ацилоїнового перетворення (рис. 4.6.3). Така перестановка Ретрон часто не очевидна недосвідченим очам.
Деякі перетворення можуть бути необхідні для захисту (ацеталів для кетонів), модифікації (зменшення кетону до алкоголю, щоб уникнути конденсації Aldol під час конденсації Claisen) або транспонувати структурний елемент, такий як стереоточка (наприклад, S N 2 інверсія, епімеризації і т.д.,) або зміщення функціональної групи. Такі перетворення не спрощують даний структурний підрозділ. Іноді активація в певних точках структури може бути введена, щоб призвести до утворення C-C зв'язку, а пізніше додаткова група може бути видалена. Наприклад, розглянемо наступний ретросинтез, в якому була введена додаткова група ефіру для полегшення ретрона Дікмана. У складних цілях комбінації таких стратегій можуть виявитися дуже продуктивною стратегією планування ретросинтезу. Свідки стратегії хімічної модифікації, наведеної нижче, для ефективного стереоспецифічного синтезу тризаміщеного олефіну (рис. 4.6.4)
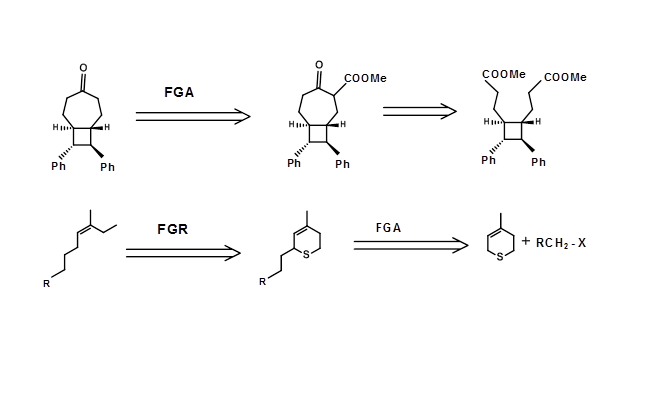
Малюнок 4.6.4 Приклади стратегій FGA/FGR для складних цілей
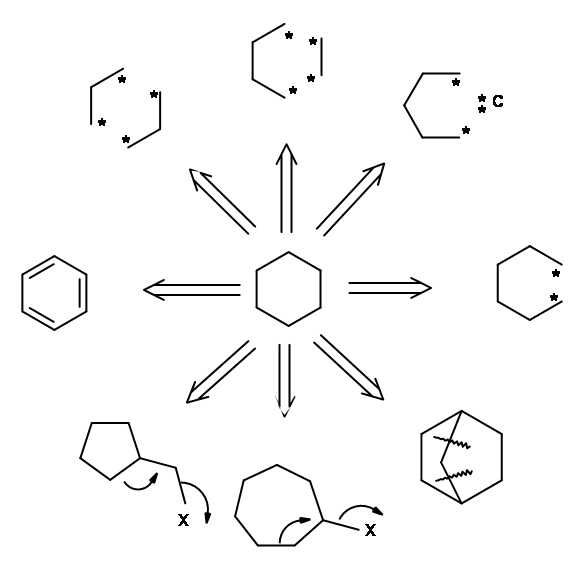
Серед молекулярних архітектур мостові кільця становлять складну проблему в процедурах відключення на основі структури. Корі запропонував керівні принципи ефективного відключення стратегічних облігацій.
Розщеплення зв'язку для ретросинтезу повинно призвести до спрощених конструкцій, бажано несучих п'яти- або шестичленні кільця. Середні і великі кільця важко синтезувати стереоспецифічно. Серед загальних кілець шестичленне кільце легко наближається і маніпулюється великим і малим кільцями. Одночасне розщеплення двох зв'язків, що передбачає циклоприєднання — ретрони часто є більш ефективними. Деякі розщеплення стратегічних облігацій показані на малюнку 4.6.5, що свідчить про хороші та погані стратегії розщеплення, засновані на цьому підході. Однак ці вказівки не є обмежувальними.
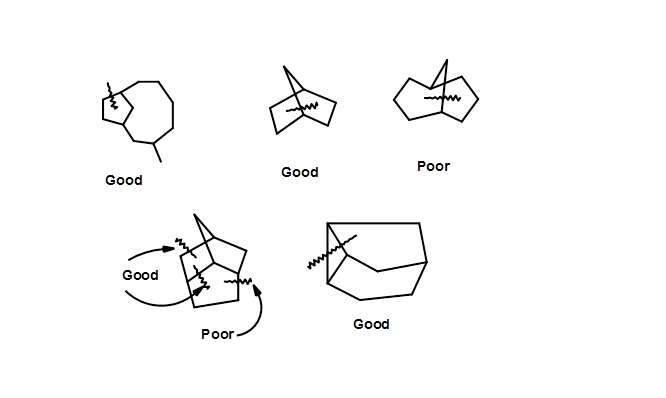
Таким чином, виявлення наборів Ретрон - перетворення в даній молекулі-мішені є критичним компонентом ретросинтезу. Такий підхід часто може генерувати кілька синтетичних маршрутів. Заслуга такого підходу в тому, що вихідні матеріали не завдають шкоди цій логіці. Розроблені таким чином ретросинтези можуть відкрити кілька маршрутів, які потребують подальшої критичної перевірки на основі відомих фактів.
Ідентифікація наборів Retron/Transforms забезпечила передумовою для комп'ютерних програм, призначених для генерації ретросинтетичних маршрутів. Список Retrons і відповідні перетворення були взаємопов'язані і дані зберігалися в комп'ютері. Таким чином, всі відомі реакції були проаналізовані на предмет їх характеристик Ретрон/Трансформації та задокументовані. Відповідні літературні цитати також були задокументовані та пов'язані. На основі цих входів були розроблені комп'ютерні програми для генерації ретросинтетичних маршрутів для будь-якої заданої структури. Кілька таких програм зараз доступні на ринку, щоб допомогти хімікам генерувати синтетичні стратегії. З огляду на будь-яку структуру, ці програми генерують кілька маршрутів. Після того, як вчений визначає конкретні маршрути, що цікавлять для подальшого аналізу, програма генерує детальні синтетичні кроки, необхідні реагенти та відповідні цитати. Незважаючи на такий потужний штучний інтелект, інтелект і інтуїтивний геній хіміка все ще здатний генерувати нову стратегію, ще не запрограмовану. Знову ж таки, людський інтелект все ще є критичним входом для аналізу маршрутів, створених за допомогою комп'ютера. Грунтуючись на досвіді команди хіміків, їх проектної мети проекту та наявних потужностей, маршрути проходять подальшу перевірку.
4.7 Опрацювання концепцій
Короткі списки синтезів, які ілюструють стратегії ретроаналізу, розроблені за допомогою потужних перетворень, наведені нижче. Кілька синтезів з хімії натуральних продуктів пізніше розглядаються в цьому розділі, що додатково ілюструє ці моменти.
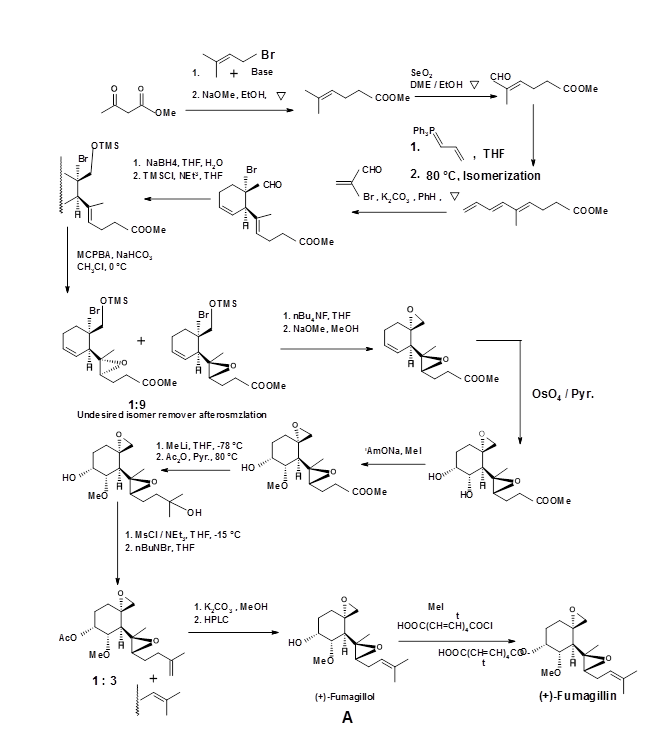
Ретросинтез на основі перетворення Дільса-Альдера; (Е.Дж. Корі та ін., J.A.C.S. (1972), 94, 2549). Фумагіллол (4.7.1A) представляє 4 стереоцентри та чутливі функціональні можливості.
Спрощення функціональних груп спочатку піддається вік-діол. Цей сайт може походити від олефіну D. Подальший ретроанліз призвів до структурно спрощеної цільової послідовності B до F. Кільцева система циклогексену підходить для потужного перетворення DA. Цей крок породив два стереоцентри в одній реакції, а також олефін у правильному положенні для гідроксилювання. Ключовий проміжний C також може бути сформований за допомогою функціональної групи перетворення, що веде до G. Це забезпечило простір для нового набору вихідних матеріалів за допомогою іншого DA Transform. Ретросинтетичний аналіз та фактичний синтез показані на малюнку 4.7.1.
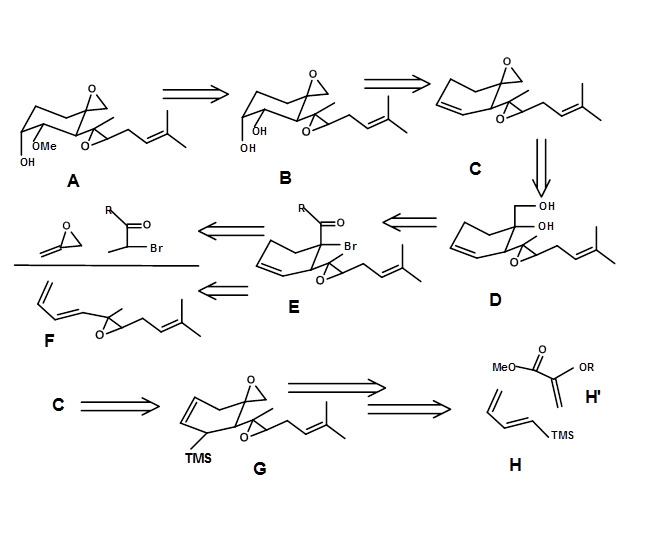
Синтетичний протокол, про який повідомляє Корі, викладено на малюнку 4.7.2
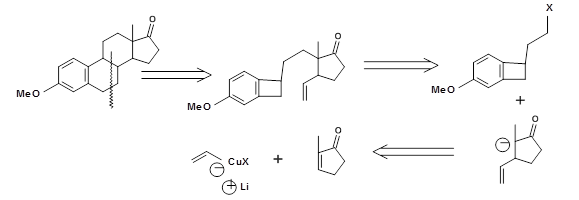
Для синтезу Естрону цікава стратегія DA Transform була розроблена Kametani et.al.. Ретросинтетическая стратегія зображена на рис. 4.7.3. Необхідний попередник дієну був сформований за допомогою реакції циклореверсії циклобутену одиниці (T. Kametani et.al., Тетраедр, (1981), 37, 3).
Важливі стереоспецифічні тризаміщені олефіни на сквалені (4.6.4B) були синтезовані за допомогою Claisen Retron 4.7.4A (рис. 4.7.4). Зверніть увагу на подвійний підхід Клейзена в цій стратегії.
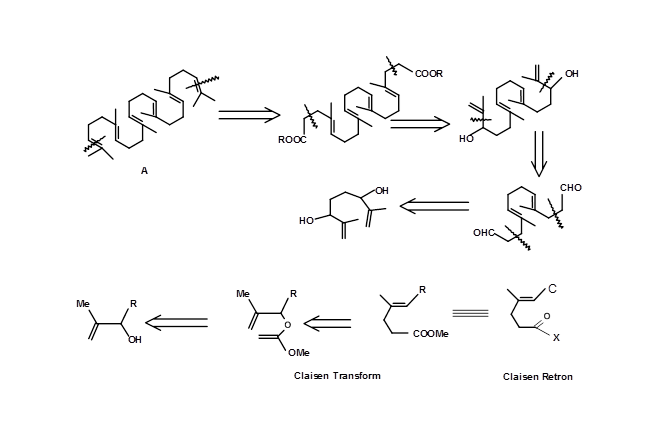
Циклізація олефінів біогенетичного типу забезпечує сферу застосування механістичного перетворення або перетворень на основі механістичних міркувань. Введення клівера хірального центру забезпечило ефективний шлях для створення декількох енантіопурних хіральних центрів за один крок, використовуючи цю стратегію (рис. 4.7.5).
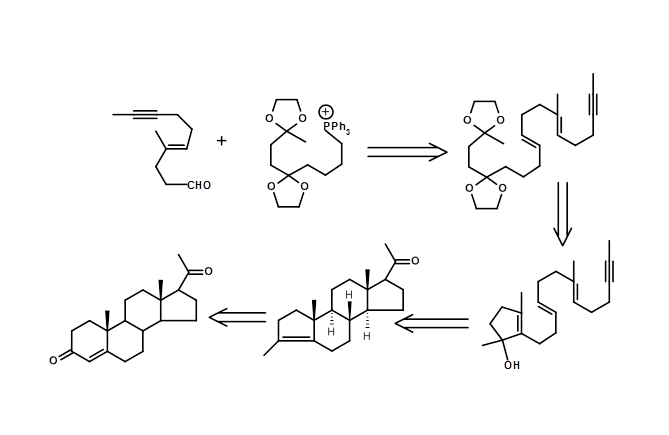
4.8 Проблема енантіомерів
У цих тривалих дискусіях вище ми дізналися про підходи до відключення. Ми сказали, що стереоцентри можуть створити особливі проблеми при плануванні ефективних синтетичних маршрутів. Давайте подивимося на молекулу біотину, щоб зрозуміти стратегії відключення та проблему стереоцентрів.
Бейкер встановив будову біотину в 1947 році шляхом однозначного синтезу молекули. Ретроаналіз синтетичної схеми наведено нижче (рис. 4.8.1).
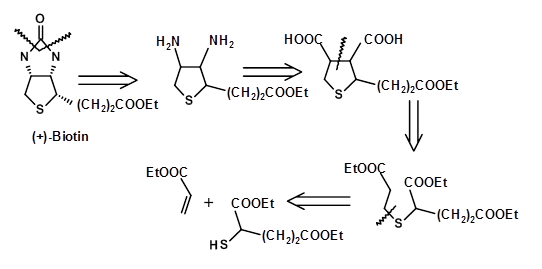
Обраний СМ і синтетичний підхід чітко встановили атомні зв'язки і загальну структуру сполуки. Однак маршрут не робив спроб синтезувати один чистий ізомер, оскільки фактична стереохімія в той час не була встановлена. Маршрут дав всі вісім стереоізомерів (3 асиметричних центру). Ці ізомери були ретельно відокремлені. У 1952 році біологічно активний ізомер був ідентифікований як весь цис-енантіомер (+) -біотоїн. На цьому етапі кілька груп повідомили про стереоспецифічний синтез всього цис-ізомеру виключно (рис. 4.8.2). Наступний ретроаналіз зображує три таких спроби. Відзначимо, що ці зусилля були спрямовані на синтез рацемата, а не чистого (+) - ізомеру біотину.
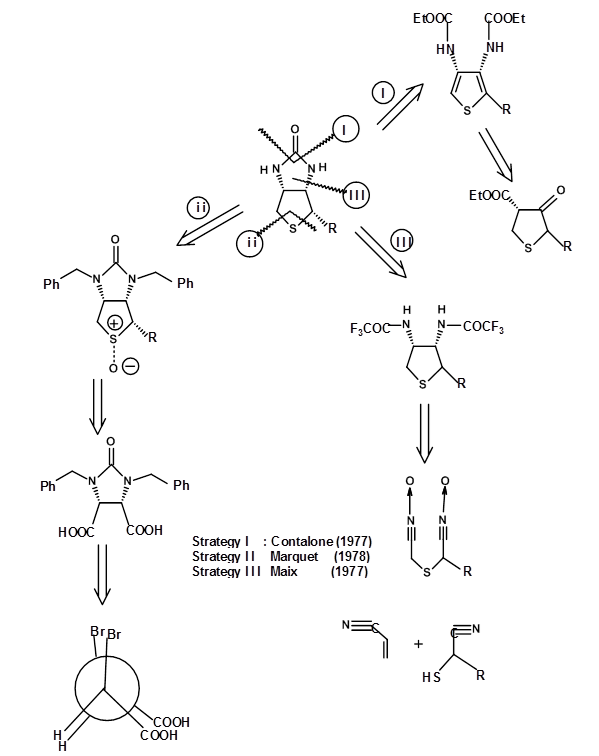
Ці підходи вирішували проблему діастереомерної чистоти. Але вони все ж залишили суміш двох нерозчинених енентіомерів а саме., (±) -біотин. Щоб отримати чистий енантіомер з чудовими врожаями, ви повинні вирішити рацемічні суміші на відповідних стадіях. По черзі можна було б вдаватися до асиметричного синтезу на всіх відповідальних етапах. Ще кращим підходом було б почати з хірального СМ, який має більшість стереоцентрів правильним чином. Цей елегантний підхід називається підходом Хірона. При ретельному виконанні такі процедури дають дуже чистий енантіомер в якості кінцевого продукту. Два таких підходи для (+) -біотину показані нижче. У першому підході вибирається хіральна амінокислота цистеїн, оскільки вона має один ключовий асиметричний центр, сірчаний фрагмент і карбонову кислоту в правильних положеннях (рис. 4.8.3). У цьому прикладі вибір СМ цілком очевидний. Відзначимо введення колуном другого азоту і етап циклізації, що веде до утворення тетрагідротіофенового кільця. Також зверніть увагу, що вихід Крам проти анти-крам (chelation) продукти можуть бути під впливом вибору реагенту. Ці види розуміння приходять лише завдяки глибокому знанню цієї конкретної реакції.
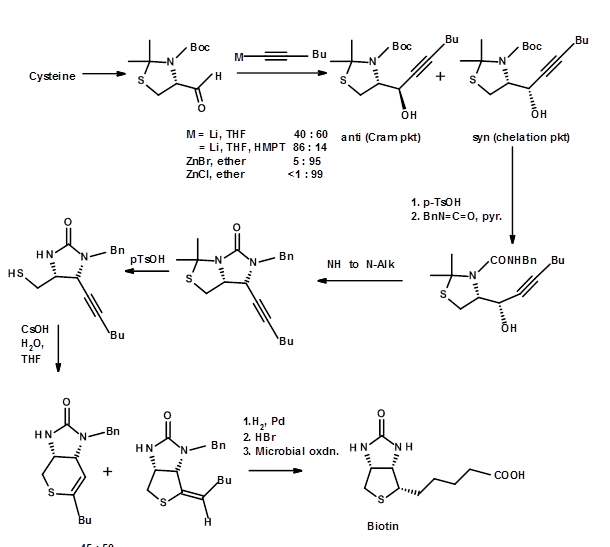
Мал. 4.8.3
У другому прикладі, обраному тут, вибір СМ як відповідного хірона не очевидний, але прихований. Такий аналіз вимагає більш критичного розуміння відповідних стереоцентрів.
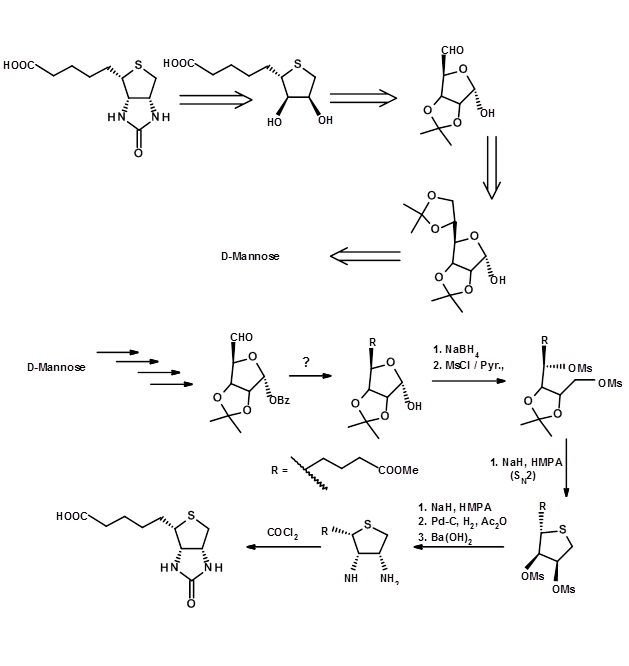
Висновок
Нова концепція в Logic In Synthesis - це навмисне планування зелених синтетичних шляхів. Логіка ретроаналізу така ж, як розглядалося вище. Єдиним відмінним моментом є те, що критерії вибору синтетичного маршруту, обговорюваного раніше, тепер аналізують те саме синтетичне дерево через вікно «Зелена хімія», щоб вибрати лише ті маршрути, які мають максимальну кількість зелених аспектів. Зелена хімія мета забезпечується шляхом включення Дванадцять принципів зеленої хімії. Це може бути зроблено шляхом використання одного або декількох з наступних методів - Використання зелених джерел енергії, таких як мікрохвильова піч, сонохімія, фотохімія і т.д., розчинник вільний синтез, використовуючи легко відновлювані нові розчинники і екологічно чисті розчинники, багаторазові каталізатори в синтезах і схемах, що дозволяють уникнути захисту групи хімія. Більшість хімії, що використовується в Green Chemistry, насправді не нова для хіміків. Хімія зараз переглядається через свідомість навколишнього середовища, яка зараз проникла в промислову хімію та суспільство в цілому. Велика частина хімії похована в двох століттях хімічної літератури. За останні роки з'явилося кілька нових відкриттів в реактивах. Тепер хіміки повинні стати більш пильними до цього пробудження до екологічних збитків, спричинених хімічною діяльністю на цьому глобусі.
Наведені вище обговорення призначені лише для ілюстрації основних кроків, пов'язаних з ретросинтетичним аналізом молекули. Ретельне знання синтетичних інструментів, механізмів та стереохімії є важливими передумовами для хіміка, щоб ризикнути в синтез складних молекул. Зайве додавати, що всі ці зусилля повинні бути належним чином підкріплені командою хіміків, маючи сувору підготовку лабораторних методів, досвід з перших рук на декількох органічних реакціях/реагентів і ретельного знання методів очищення, спектроскопічних методів і не в останню чергу, хороший знання методів пошуку для сканування та отримання необхідної інформації з величезної хімічної літератури, накопиченої з самого початку сучасної хімії.
Ретроаналіз деяких цікавих молекул
Давайте тепер зупинимося глибоко в декількох вибраних структурах, обраних з природної хімії продуктів, і подивимося, як ці структури були вирішені за допомогою різних синтетичних стратегій. Ми б почали з простої молекули - Disparlure - лише з двома асиметричними центрами. Курс закінчиться ароматом деяких зелених синтезів на основі хімії, щоб привернути увагу студентів до цього нових концепцій і проблем.