5.2: Гризеофульвін
- Page ID
- 18720

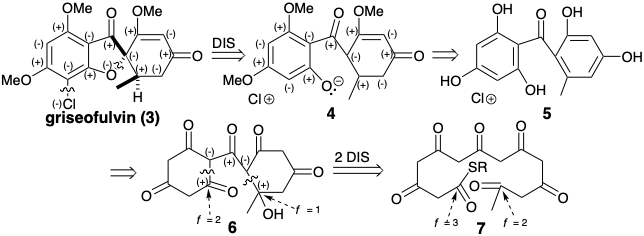
Аналіз полярної реактивності гризеофульвіну (3) виявляє дисонансні схеми за участю хлорозамінника та фуранового кисню. Дислокація, що розщеплює ці дисонансні схеми, передбачає цілком приголосний попередник 4 або ароматичний близький родич 5. Це відключення - між загальним атомом, спіро-вуглецем і не загальним атомом, фурановим киснем - також призводить до значного топологічного спрощення. Роз'єднанню кільцевого кільця в 5 для утворення ациклічного попередника має передувати перетворення кільцевих зв'язків C = C в C-C зв'язки. Таким чином, додавання води або тавтомеризація дає попередник 6, в якому можливе полярне відключення одиночних зв'язків С-С. Рівень функціональності в електрофільних центрах в ациклічному попереднику 7, запропонований цим полярним роз'єднанням, повинен бути на одну одиницю вище, ніж в проміжних 6, якщо циклізація 6 повинна давати 7 безпосередньо.
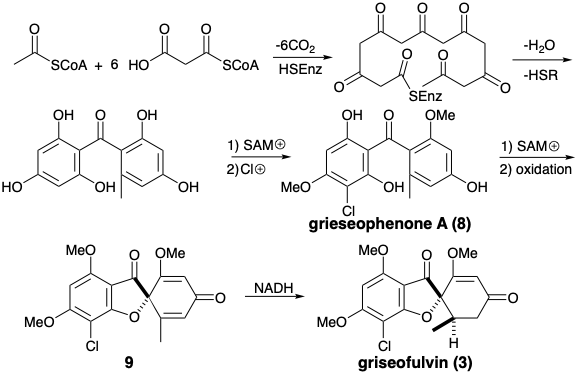
Біосинтез гризеофульвіну (3) ілюструє участь електрофільного ароматичного заміщення та окислювального зв'язку в перетворенні полі-β-кетометиленового ланцюга в функціонально і скелетно-складний ацетогенін. Так, внутрішньомолекулярні альдольні конденсації тіоефіру пов'язаного з ферментом 3,5,7,9,11,13-гексакетогексадеканової кислоти з подальшим гідролізом, О-метилуванням S-аденозилметіоніном (SAM) і електрофільним ароматичним хлоруванням утворюють проміжний продукт, гризеофенон А (8). Дисонансне з'єднання в фурановому кільці гризеофульвіна створюється окислювальною зв'язкою, яка генерує дегідрогризеофульвін (9) з 8. Стерео-селективне зменшення 9 з NADH потім доставляє гризеофульвін (3).
Біоміметичний синтез гризеофульвіну
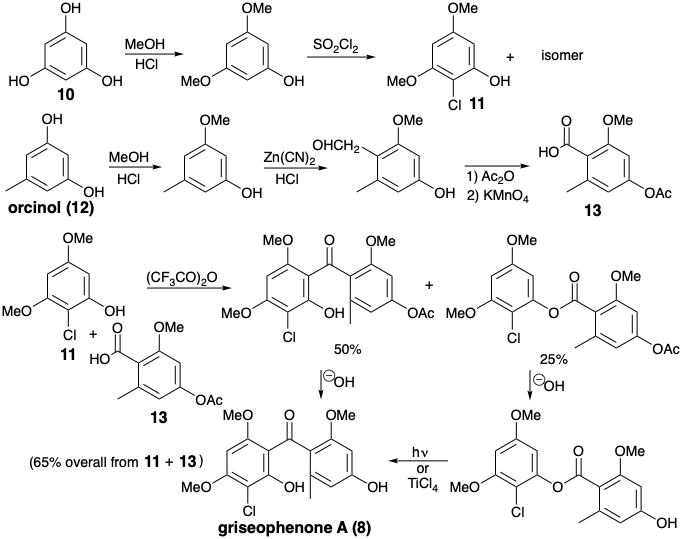
Феррицианідно-індуковане окислювальне з'єднання гризеофенону А (8) з дегідрогризеофульвіном (9) з використовували в біоміметичному тотальному синтезі рацемічного гризеофульвіну. 2 Симетричні вихідні матеріали, флороглюцинол (10) та орцинол (12) були розроблені в проміжні продукти 11 та 13 шляхом добре прецедентних електрофільних ароматичних заміщень. Ацилювання 11 з 13 відбувалося в основному при вуглеці. Продукт O-ацилювання був легко перебудований на продукт С-ацилювання, тим самим забезпечуючи гризеофенон A (8) хороший загальний вихід. Перетворення 8 в d, l-гризеофульвін (3) тісно паралельно біосинтетичний шлях.
Стратегії відпалювання Полярного циклогексенону для гризеофульвіну
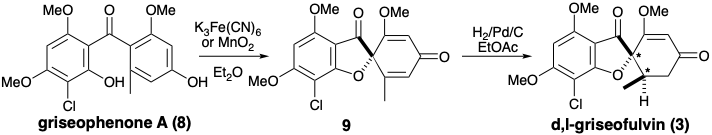
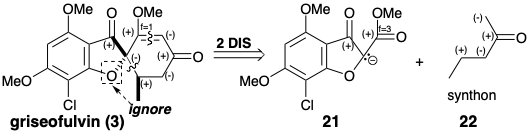
Стратегія загального синтезу гризеофульвіну запропонована полярним аналізом, який ігнорує полярну активацію, що забезпечується фурановим киснем. Відключення двох зв'язків із загальним атомом, спіро-вуглецем, у 3 призводить до значного топологічного спрощення та передбачає нуклеофільний попередник синтону 14 та біелектрофільний попередник синтон 15. Eneyne 16 - це синтетичний еквівалент 15, який повинен забезпечити 3 безпосередньо, оскільки 1,4-додавання нуклеофіла зменшить рівень ненасичення кожного електрофільного центру на одну одиницю.
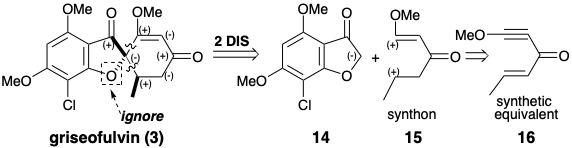
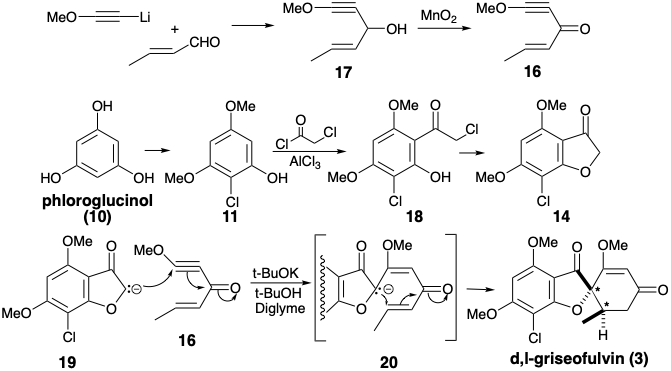
Біелектрофіл 16 готували з метоксіацетилену та кротональдегіду, а попередник 14 необхідного дисонантного нуклеофілу був отриманий з флороглюцинолу (10) через 18, отриманого ацилюванням 11 хлорацетилхлоридом. Слід зазначити, що дисонанс в фурановому кільці 14 походить від дисонантного попередника хлорацетилхлориду. Обробка суміші 14 і 16 з основою подається 3 за допомогою аніонів 19 і 20. 3
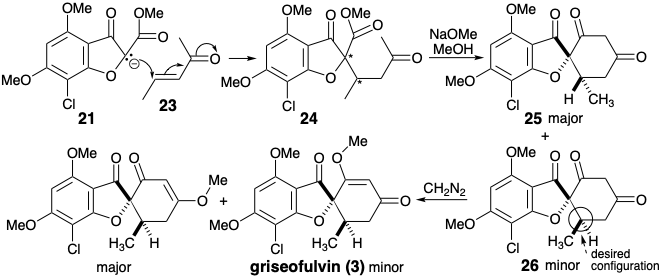
Інша стратегія також пропонується полярним аналізом, який ігнорує полярну активацію, що забезпечується фурановим киснем. Відключення двох зв'язків циклогексенонового кільця, як у 3, свідчить про добре прецедентну відпалювання циклогександіонів, яка схожа на відпалювання Робінзона (див. Розділ 4.7). На відміну від попередньої стратегії, метиловий ефір не виробляється безпосередньо. Енон 23 служить синтетичним еквівалентом для синтетону 22.
Конденсація 21 з 23 дає два діастереомерних циклогександіони 25 і 26. Цей синтез менш ефективний, ніж попередній, оскільки основний діастереомер 25 є епімерним з природним продуктом 3, а метілетерифікація незначного діастереомеру 26 відбувається з несприятливою регіоселективністю. 4
Стратегія циклододавання для гризеофульвіну
Стратегія Данішефського для загального синтезу гризеофульвіну 5 була розроблена навколо його методу відпалу циклогексенону через реакцію Дільса-Альдера висококисневих дієнів, наприклад, 27.
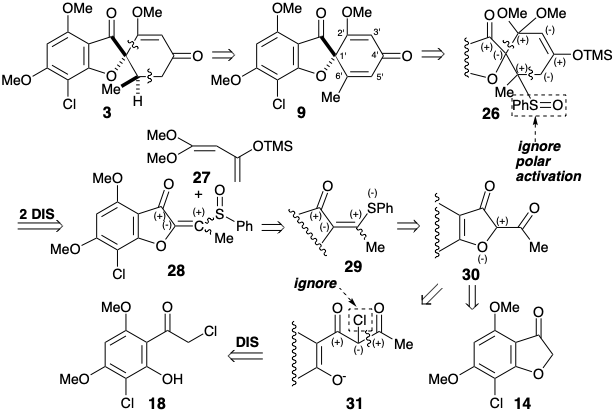
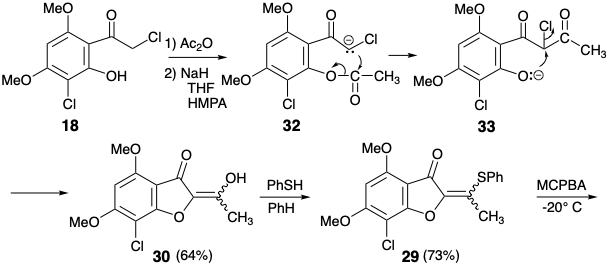
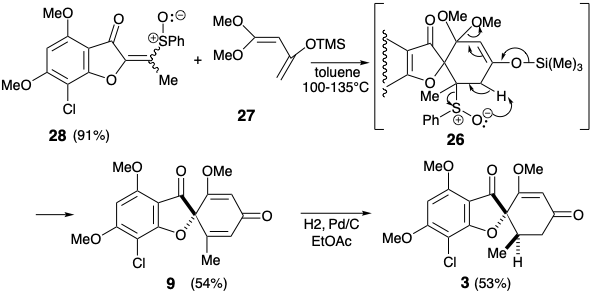
Ця гранична умова направляє та направляє ретросинтетичний аналіз для пошуку двозв'язкового відключення кільця циклогексенону до попередника бутадієну та дієнофілу. Крім того, стереоселективне перетворення 9 до дати 3 встановлених раніше 2 було прийнято для спрощення стереохімії цілі. Вивих до 9 видаляє один асиметричний центр, який може бути введений в кінці синтезу шляхом стереоселективного гідрування. Для бажаної реакції Дільса-Альдера потрібен зв'язок C = C між вуглецями 3' і 4'. Таким чином, облігації 2 '3' і 5'6' C = C в 9 повинні бути сформовані після ключового кроку Дільса-Альдера. Реквізит 3 '4' C=C зв'язок забезпечується дислокацією 9 до похідної енол-кета1 26. Облігація 5 '6' C = C може бути введена в 26 за допомогою різних процесів ліквідації. Вибір сульфоксиду як вихідної групи продиктований додатковою корисністю групи сульфоксидів для активації діенофіла 28 до реакції Дільса-Альдера з дієном 27, який обов'язково багатий електроном. Також можна очікувати, що сульфоксид не контролюватиме структурну селективність реакції Дільса-Альдера, яка буде контролюватися замість цього карбонільною групою фуранонового кільця в 28. Електрон, що виводить сульфоксид група дисонансна по відношенню до фуранону карбонілу в 28, але її можна отримати шляхом окислення відповідної електронної донорської сульфідної групи в 29. Цей приголосний вініловий сульфід є просто похідним сульфіду енолу попередника 30. Цей діон може бути доступний шляхом ацилювання фуранону 14, який використовувався в попередньому синтезі гризеофульвіну. Крім того, будівництво дисонансного зв'язку C-O в 30 може бути досягнуто після завершення вуглецевого скелета, але нуклеофуга буде потрібно в 31, оскільки карбонільні групи не можуть забезпечити необхідну електрофільність. Ігноруючи хлорну групу в 31, 1,3-дикарбонільний масив є приголосним і може бути побудований за допомогою Claisen ацилювання кетону 18, який також використовувався в попередньому стереоселективному синтезі, розглянутому вище.
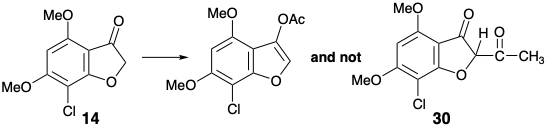
Насправді O-ацилювання аніону енолату з 14 відбувається до повного виключення ацилювання вуглецю, необхідного для отримання 30. З іншого боку, внутрішньомолекулярна доставка ацетильного електрофілу в 32 служила для розкриття внутрішньомолекулярного O-алкілування проміжного фенолату 33 доставив бажаний діон 30, який існує як еноловий таутомер. Перетворення енолу сульфіду 29 з 30 у відповідний сульфоксид супроводжувалося селективним окисленням сульфіду в присутності зв'язку C = C з MCPBA при низькій температурі. Додавання Дільса-Альдера отриманого вінілсульфоксиду 28 до 1,3-дієну 27 супроводжувалося термічною елімінацією фенілсульфенової кислоти та метокситриметилсилану з проміжного циклогексену 26 для доставки циклогексадіенону 9.