5.3: Тетрацикліни
- Page ID
- 18721

Ацетил CoA - не єдиний тіоефір, який може ініціювати олігомеризацію ферментного матриксу малонілу КоА. Так, наприклад, був постульований біосинтез для тетрацикліну (44), що включає конденсацію малонамоїлу CoA (34) з вісьмома молекулами малонілу CoA з отриманням зв'язаного ферментом полікетоаміду тіоефіру 35, який частково дегідроциклізується після метилювання одного метилен і відновлення одного карбонілу. 6 Кінцеве кільце тетрациклінової кільцевої системи утворюється циклізацією Дікмана від 36 до 37 після вивільнення частково циклізованого полікетиду з полікетидсинтетази. Два ароматичних гідроксилювання підвищують функціональність після завершення вуглецевого скелета. Ці гідроксиляції можуть включати проміжні оксиди арени, наприклад 37 → 38 → 39. Редуктивне амінування 40 через 41 і 42 до виходу 43 супроводжується окислювальною дезамінацією глутамінової кислоти.
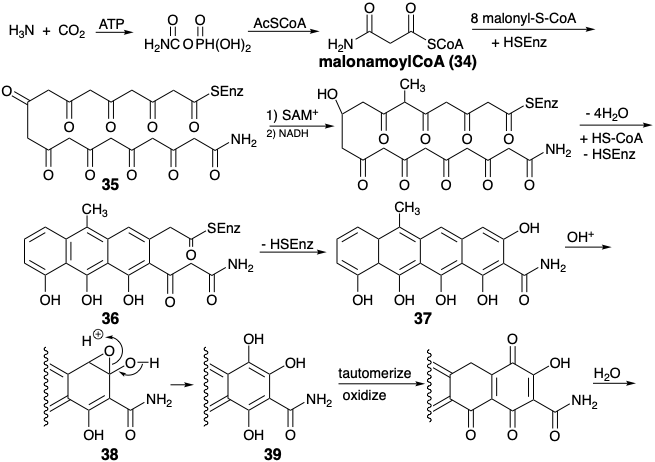
Топологічний аналіз плавлених кільцевих систем. 7
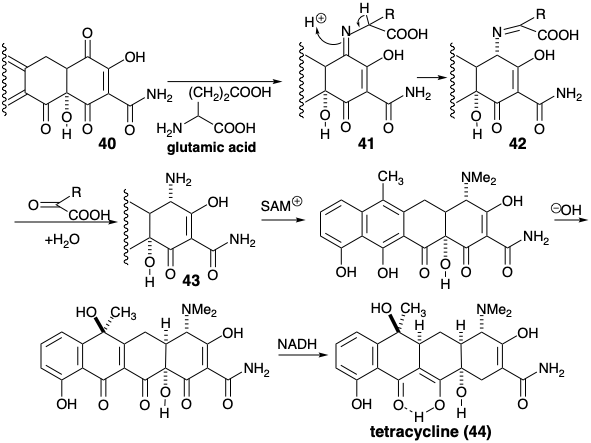
Кільцева пара - це система плавленого кільця, якщо два кільця поділяють один і лише один загальний зв'язок, злитий зв'язок. Стероїдні та тетрациклінові кільцеві системи є прикладами багатоциклічних структур, що містять лише пари плавленого кільця, на відміну від спіроциклічних або мостовидно-кільцевих пар. Крім злитих зв'язків (позначені f), діаграми нижче вказують загальні атоми (показані як •), і ексендозв'язки (позначені e), які є exo до одного кільця і ендо іншого, і зв'язки (позначені пунктирними лініями), які утворюються під час біосинтезу цих кільцевих систем з ациклічні попередники.
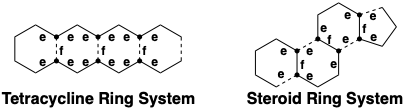
Існує цікавий контраст між топологічними стратегіями тетрациклінового і стероїдного біосинтезу. Обидві стратегії включають ключові ациклічні проміжні продукти, які включають всі атоми скелетного вуглецю. Однак біосинтез тетрациклінового скелета передбачає формування всіх кільцевих злитих зв'язків, тоді як при біосинтезі стероїдного скелета утворюються тільки ексендозв'язки. Інший контраст зустрічається в біосинтетичної генерації периферійного кільця, тобто кільця, яке залишається після розщеплення всіх злитих зв'язків. Таким чином, під час біосинтезу тетрациклінового скелета генерується лише одна периферична зв'язок, тоді як чотири зв'язки периферичного кільця генеруються при біосинтезі стероїдів.
Також примітним є той факт, що в обох біосинтезах зв'язки між парами спільних атомів є стратегічним зв'язком, тобто стратегічно важливим для досягнення швидкого зниження молекулярної складності шляхом відключення під час дислокації синтетичної мішені. Дислокації стратегії біосинтезу тетрацикліну рекомендуються як топологічним, так і полярним аналізом реактивності. Топологічно стратегія роз'єднує всі зв'язки між парами спільних атомів. Аналіз полярної реактивності виявляє достатню функціональність. Якщо активація, що забезпечується декількома функціональними групами, ігнорується, численні функціональні групи залишаються виключно приголосними сполучними ланцюгами, які можуть бути створені шляхом використання цільової функціональності в попередниках у поєднанні з декількома доданими приголосними функціональними (карбонільними) групами.
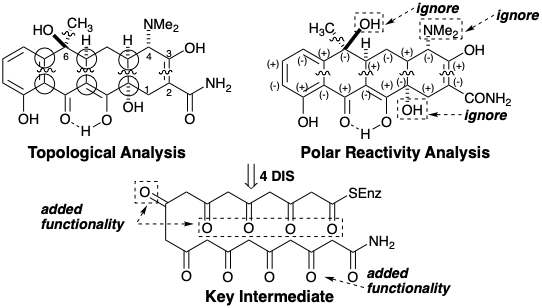
Лінійна стратегія для тетрациклінів
Перший синтез біологічно активного (хоча і неприродного) похідного тетрацикліну був досягнутий Вудвордом та колабораторами.8 Ці працівники спростили синтетичну мету, не включивши лабільний третинний 6-гідроксил, а також 6-метил тетрацикліну (44). Кільце А 45 є функціонально і стереохімічно найскладнішою частиною цієї спрощеної мети. Це кільце настільки високо функціоналізовано, що полярний аналіз неоднозначний. Він містить безліч дисонансів полярної реактивності. Якщо це кільце відірвати від 45, приголомшливе спрощення синтетичної мішені результатів. Не тільки видаляється велика кількість реактивної функціональності, але й реалізується топологічне спрощення. Таким чином, шляхом розщеплення пари ексендозв'язків, які є віцинальними і коциклічними (в одному і тому ж первинному кільці, тобто тієї, яка не розрізана на пару менших кілець трансанулярним містком) видаляються всі залишки А-кільця. Решта синтепон BCD є відносно хімічно стійким, структурно простим фрагментом 46. Можливий синтетичний проміжний продукт, тобто належним чином функціоналізований молекулярний фрагмент, що відповідає 46, дорівнює 47. Карбонільна група в кільці В 47 забезпечує активацію для вироблення кільця А. Метиловий ефір в кільці D блокує депротонацію фенолу. Полярний аналіз неароматичної частини 47 передбачає дислокацію до 48 і диметилоксалат, симетричний дисонантний біелектрофіл. Відпалювання 48 шляхом ацилювання Friedel-Crafts 49 буде використовувати нуклеофільну реакційну здатність ароматичного D-кільця в 49, що активується цільовою метокси-групою, і електрофільною реакційною здатністю карбонілу.
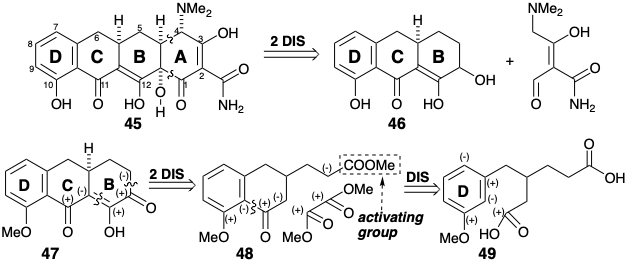
Однак ця стратегія є потенційно недосконалою, оскільки електрофільна заміщення, яка повинна відбутися орто до метокси-замінника в кільці D 49 під час перетворення на 48, також може відбуватися в нуклеофільному положенні пункт до активуючої метокси-групи. Щоб виключити параацилювання, хлорозамінник у попереднику 50 може бути використаний як блокуюча група (замінник, введений для контролю реактивності та згодом видалений).
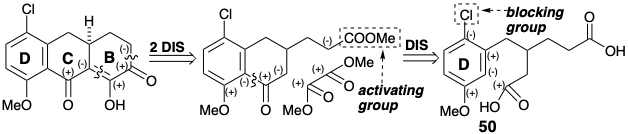
Топологічно дислокація від 49 до 51 + 52 бажана, оскільки з ароматичного кільця видаляються всі залишки бічного ланцюга. Однак полярний аналіз 49 показує, що електрофільне алкілювання або ацилювання попередника анізолу сприятиме заміщенню орто або пара, а не метазаміщення, необхідному для генерації 49.
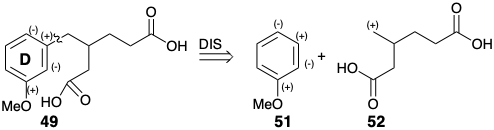
Дислокація 49 ⇒ 53 + 54 рекомендується готовою наявністю бензилових органометаліків, відповідних 53. Нуклеофільний синтон, такий як 53, може дозволити собі діефір 49 на 1,4-додавання до 54. Насправді відомий загальний синтез β-заміщених алкандіових кислот, таких як 49, який пов'язаний з цим підходом. 9 Дислокація до більш зв'язаних, циклічних напівпродуктів β-кетоефіру 55 виявляє можливу корисність легкодоступних попередників циклоалкенону для синтезу β-заміщених алкандіових кислот шляхом ретро-розщеплення Дікмана.
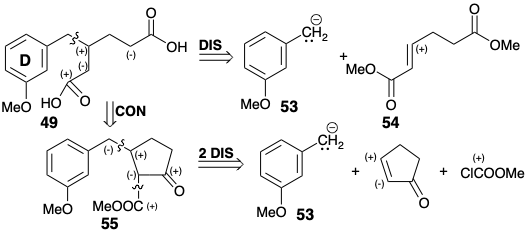
На практиці С-ацилювання проміжних продуктів енолатів, таких як 56, супроводжується O-ацилюванням одержуваних β-кето-ефірів. Однак ніяких додаткових кроків не потрібно, оскільки отримані ефіри енолу гідролізуються до β-кето-ефірів в умовах реакції, необхідних для ретро-розщеплення Декмана останнього для генерації цільових β-заміщених алканових діоєвих кислот.
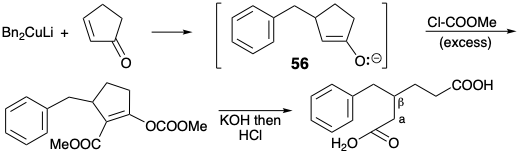
Замість бензилового нуклеофіла в якості вихідного матеріалу стратегія Вудворда базувалася на виборі легкодоступного бензилового електрофілу, метил-м-анізату (57), в якості вихідного матеріалу. Цей вибір каналів ретросинтетичного аналізу попередника 58 з активуючої функціональної групи, бензиловий карбоніл, який повинен бути видалений згодом, щоб забезпечити 49. Хоча кон'югація з віддаленим карбометоксилом у 58 може забезпечити нуклеофільну активацію, необхідну для об'єднання вихідного матеріалу гександіового ефіру з 57, Вудворд вирішив використовувати класичний синтетичний метод кетонів, алкілювання β-кето-ефіру з подальшим гідролізом і декарбоксилювання (синтез ацетооцтового ефіру), щоб зібрати вуглецевий скелет 49. Цей вибір передбачає включення групи активації карбонового ефіру в 58.
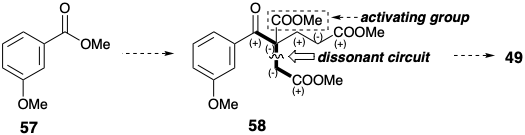
Вудворд вивчив три різні стратегії, щоб виявити, який маршрут насправді забезпечує найкращі загальні врожаї 58 з 57. Кожен шлях використовує легкодоступні вихідні матеріали. Цікаво, що більш тривалий (менш конвергентний) маршрут, використовуючи метилацетат і метилхлорацетат як будівельні блоки, дав кращі загальні врожаї, ніж інші два шляхи, які використовують симетричні попередники діефіру. Також важливо, що в кожному маршруті використовується дисонансний вихідний матеріал для забезпечення дисонансного контуру в 58.
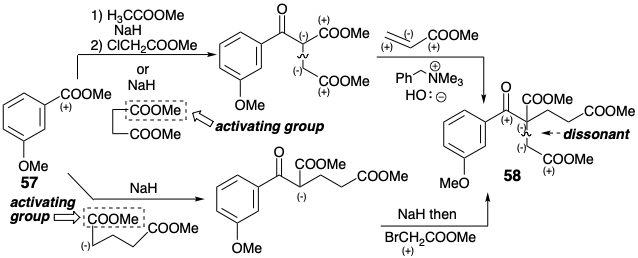
Перетворення 58 в ключовий трициклічний проміжний 61 був досягнутий шляхом гідролізу, декарбоксилювання, гідрогенолізу, хлорування, ацилювання Фріделя-Crafts, Claisen конденсації-циклізації Dieckmann, а потім ще один гідроліз і декарбоксилювання. Цікаве селективне деметилювання, яке отримало 60, є результатом внутрішньомолекулярної переетерифікації проміжного бензилового спирту з подальшим гідрогенолізом отриманого бензилового ефіру. Метилювання 60 проводилося виключно для полегшення очищення.
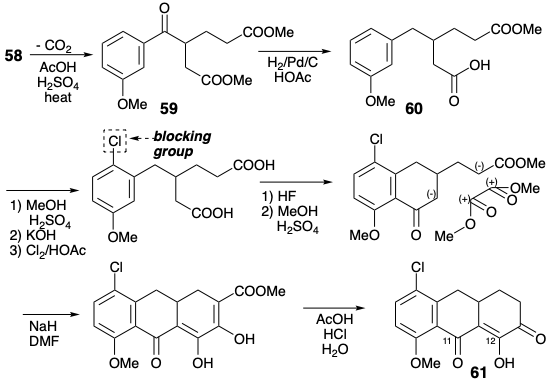
Вудворд передбачав різні властивості для трьох карбонільних груп в 61. Таким чином, β-дикарбонільний масив (C-11 і 12) є «стабілізованою системою вінілової карбонової кислоти, тоді як третя, як і в простих α-кето-кислотах, повинна бути як сильно сприйнятливою до реакцій додавання, так і легко енолізуватися. Остання властивість повинна надавати високу нуклеофільну реакційну здатність сусідньому метилену». Ця реакційна здатність була використана для додавання до 61 фрагмента попередника для кільця А, а потім диметиламіно групи полярними реакціями. Третій карбоніл, виконуючи свою мету, потім видаляли шляхом відновлення до α-гідрокси-кетону 63, активації шляхом внутрішньомолекулярної переетерифікації та відновного розщеплення отриманого лактону.
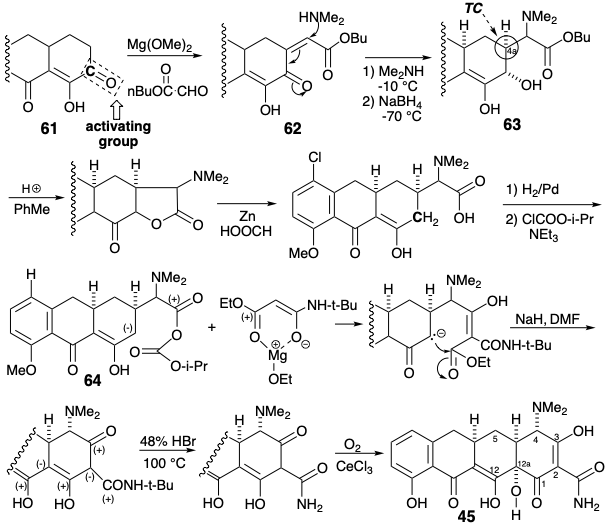
Потім був розроблений повністю функціоналізований ациклічний попередник для кільця А шляхом ацилювання метилмалонаматного карбонамату зі змішаним анігідридом 64. Циклізація Дікмана, що використовує нуклеофільну реакційну здатність, що надається сусідньому метилену карбонілом в положенні 12, виробляється кільце А. Необхідна стереохімія в положенні 4а в 64 виробляється при додаванні диметиламіну до 62, що сприяє більш стабільному екваторіальний епімер 63. Встановлення останньої функціональної групи, гідроксилу на позиції 12а потім було здійснено окислювальним шляхом, щоб доставити 45.
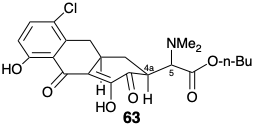
Більш конвергентна стратегія для тетрациклінів.
В іншій стратегії загального синтезу 6-дезметил-6-дезокситетрацикліну (45), як і в попередньому синтезі Вудвордом, мішень спрощується шляхом видалення гідроксилу С-12а. Тоді, за винятком полярної активації, що забезпечується C-4 диметиламінозамісником, полярна реактивність, що забезпечується рештою функціональних груп, є повністю приголосною вздовж будь-якого контуру, як показано в 65. Полярне відключення 65 ⇒ 66 розділяє молекулу на два великих фрагмента, об'єднаних простим метиленовим мостом. Подальші роз'єднання зв'язків роз'єднують кільце А на два прямоланцюгові попередники 67 і 68, які можуть возз'єднатися полярними реакціями. Синтетичні еквіваленти, які Муксфельдт використовував для синтонів 67 та 68, були 69 та 70a відповідно. 9
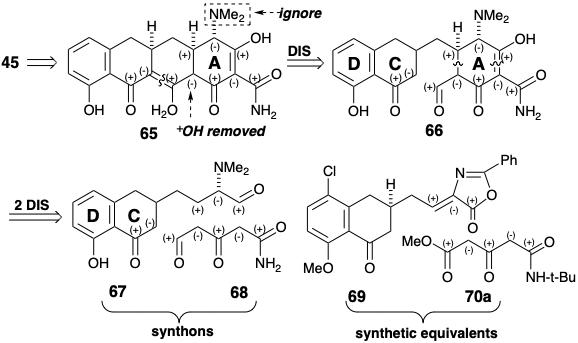
Родзинкою синтезу Муксфельдта є геніальна реакція, яка виробляє тетрациклічний проміжний продукт 71 за один крок від біциклічного попередника 69 у виході 82%! Остаточне регулювання функціоналу з готовністю дозволяє 45 з 71. Стратегія виграє від високого ступеня конвергенції. Більше того, 69 легко доступний синтезом азалактону з 72 і 73. Нарешті, проміжний CD-кільцевий альдегід 72 був отриманий з хорошим загальним виходом з 4-хлор-3-метиланізолу.
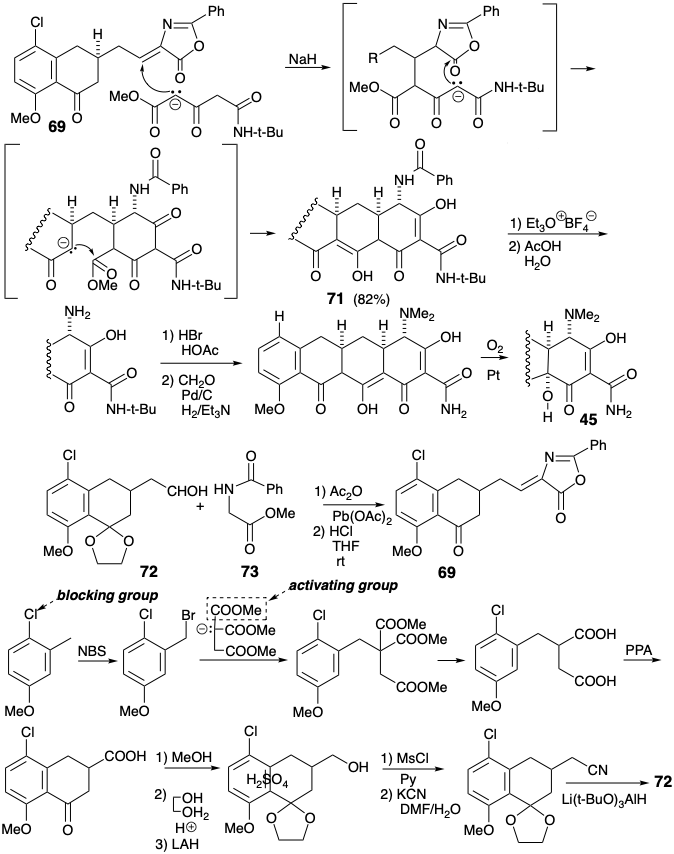
Перший загальний синтез природного тетрацикліну, 5-гідроксипохідного терраміцину, був досягнутий Muxfeldt. 10 Кільця A і B були зібрані за тією ж стратегією, яка використовувалася вище для генерації 45. Так, CD-кільцевий альдегід 74, аналогічний 72, був конденсований з попередньо сформованим азатіолактоном 75, аналогічним проміжному азалактону, який бере участь в реакції 73 з 72. Отриманий Michael акцептор 76, аналогічний 69, потім був конденсований з незахищеним амідом 70b, відповідним 70a, щоб генерувати тетрациклінову кільцеву систему в одному синтетичному етапі. Подальша депротекція маскованих гідроксилів та окислювальне введення останнього гідроксилу в положенні 12а супроводжувалося видаленням маскувальної групи тіобензоїлу в виключно м'яких умовах після обробки йодидом метилу. Ця реакція включає в себе генерацію і подальший гідроліз метилтіомідного ефіру проміжного продукту. Нарешті, контрольоване диметилювання первинного аміну дало терраміцин.
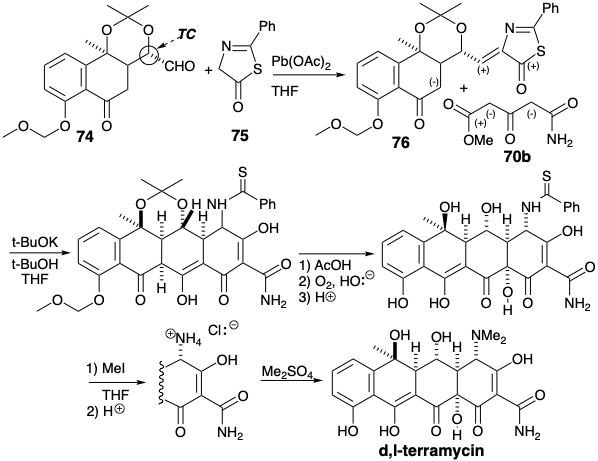
Стратегія, що використовується для генерації ключового проміжного (74), використовує тимчасовий міст у 78, щоб маскувати альдегід у прихованій формі як алкен та створити складчастий трициклічний попередник, який, як очікується, додасть метиловий нуклеофіл до карбонілу в положенні 6 від найменшого стерично перевантажене опукле обличчя. Це забезпечує цис-зв'язок між метильною групою та сусіднім протоном плацдарму. Очікується, що вінілогічний α-дикетоновий масив у 77 буде особливо електрофільним, достатньо активованим, що конкуренції від додавання до ацетатного карбонілу можна уникнути. Селективність, що сприяє додаванню до карбонілу в положенні 6, а не 11, може бути наслідком зниження електрофільності внаслідок кон'югації 11- але не 6-карбонілу з киснем у положенні 10. Наявність циклогексену в 77 рекомендує синтез циклоприєднання, що включає подвійно активований електронно-дефіцитний хінон-діенофіл і відносно багатий електронами 1-ацетокси-1,3-бутадієн.
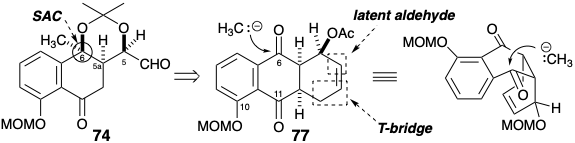
Насправді реакція Дільса-Альдера ацетоксибутадієну з ацетатом юглону (78) дала діацетат 79 регіоселективно, а додавання метильного реагенту Гріньяра до 79 є регіо- та стереоселективним. Циклогексенове кільце, виконуючи свою мету, потім окислювалося, щоб утворити альдегід з його прихованого еквівалента - подвійного зв'язку вуглецево-вуглецевого. Потім непотрібний двовуглецевий фрагмент був видалений послідовністю, що включає конденсацію альдолу, окислювальне розщеплення і, нарешті, ретро розщеплення Клайсена проміжного β-дикетонового масиву в 80.
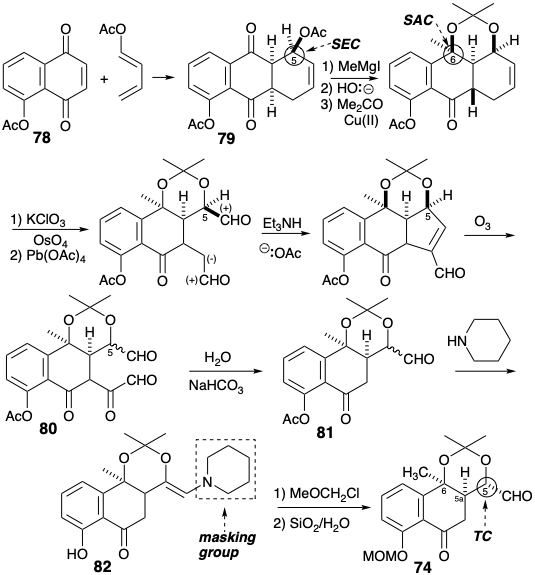
Через стереоелектронну перевагу ендо перехідного стану в реакції Дільса-Альдера 78 генерується 79 з правильною конфігурацією в положенні 5. Однак окислювальне розщеплення, яке генерує 80, виробляє суміш епімерів у положенні 5. Під час заміни ацетилзахисної групи метоксиметилом піперидин використовується як нуклеофіл для розщеплення ацетату і як маскує група для приховування чутливого альдегіду під час алкілування фенолу. На щастя, гідроліз енаміну 82, шляхом обробки вологим силікагелем, генерується епімерно чистий 74 з необхідною конфігурацією в положенні 5.