8.9: Мініпреп ДНК шляхом лужного лізису (активність)
- Page ID
- 6498

лужний лізис
Після того, як ДНК впроваджується та переноситься бактеріями, ми хотіли б знову ізолювати ДНК для подальших маніпуляцій. Для цього бактерії, що містять цікаву плазміду, вирощують у рідкій культурі багатому поживними речовинами бульйоні, виготовленому з дріжджового екстракту під назвою бульйон Лурія-Бертані (LB). Ці культивовані бактерії вирощуються до тих пір, поки вони не мають високої концентрації протягом ночі. Їх збирають через центрифугування і відвар видаляють. Отримана гранула бактерій ресуспендується в фізіологічному буфері, що містить хелатор ЕДТА. Хелатор - це хімічна речовина, яка видаляє двовалентні катіони, як Ca 2+ або Mg 2+ з розчину. Це важливо, оскільки двовалентні катіони необхідні для того, щоб ферменти перетравлювали ДНК, щоб бути активними. Хелатируючи іони, ДНК, яку ми зрештою хочемо очистити, буде безпечною від деградації.
Після ресуспензії бактерій в бактеріальну суміш змішують лужний розчин 0,1N NaOH. Цей розчин також містить іонний миючий засіб під назвою додецилсульфат натрію (SDS), який допомагає денатурувати білки та порушувати їх взаємодію з ДНК. Суміш стає в'язкою в міру того, як бактерії лопаються і їх вміст витікає в розчин. Цей основний розчин потім нейтралізується буфером ацетату калію при рН 5,5. Коли розчини змішуються разом, рН наближається до 7, а калій взаємодіє з SDS, щоб викликати осадження геномної хромосомної ДНК та білків. Для того, щоб відокремити осад від розчину, суміш центрифугують на високій швидкості, щоб гранулювати геномну ДНК і білок. Супернатант, або розчин, переноситься в колону, що містить кремнезему мембрану. В умовах високої солі ДНК прилипає до скла або кремнезему. Пропускаючи розчин через цю колонку, плазмідна ДНК в супернатанті потрапляє на кремнезему мембрану і видаляється з розчину. Додаткові змивки використовуються для видалення бродячих забруднень і видалення зайвої солі. Плазмідна ДНК остаточно видаляється з колони шляхом елюції низьким буфером солі. Цей буфер з низьким вмістом солі - трис рН 8 з ЕДТА (TE). Плазмідна ДНК може стабільно зберігатися в буфері TE в морозильній камері протягом тривалого періоду часу.
Вправа 1: Міні-підготовка плазмідної ДНК методом лужного лізису
- Прищепити 2 мл насиченого середовища (LB, YT або приголомшливий бульйон), що містить відповідний антибіотик з однією колонією перетворених бактерій. Інкубують культуру протягом ночі при 37° C з енергійним струшуванням. (Це те, що вам надали)
- На кожну групу слід взяти по 2 культури.
- Центрифугуйте культуральні пробірки безпосередньо максимум протягом 5 хвилин.
- Якщо не здатні крутитися в цих тюбиках, перенесіть 1,5 мл культури в мікрофужну пробірку (Еппендорфський тюбик).
- Центрифуга на максимальній швидкості протягом 30 сек.
- Коли центрифугування буде завершено, влийте розчин відвару в ємність з хлорним вапном.
- Ресуспендуйте бактеріальну гранулу в 250 мкл крижаного розчину Р1 енергійним струшуванням і перенесіть назад у мікроцентрифужну трубку.
- Р1 - фізіологічний розчин 50 мм Тріс при рН 8.
- P1 містить хелатор називається ЕДТА.
- Хелатори зв'язують надлишок двовалентних катіонів, які необхідні для активності ДНКАзи.
- Ліза: Додайте 250 мкл розчину Р2 до кожної бактеріальної суспензії. Щільно закрийте трубочку, і перемішайте вміст, перевернувши трубочку акуратно п'ять разів. Чи не вихори! Зберігайте трубочку на льоду.
- Це буфер лізису, що містить миючий засіб Додецилсульфат натрію та NaOH.
- Нейтралізувати : Додайте 350 мкл крижаного розчину Р3. Закрийте трубку і розігніть розчин лізису, перевернувши трубку кілька разів. Зберігати трубочку на льоду 3-5 хвилин.
- Це нейтралізаційний буфер, що містить ацетат калію.
- Нейтралізація відновлює рН майже до 7, а також спричиняє осадження геномної ДНК та білків у похмурий безлад (соплячий).
- Центрифугуйте бактеріальний лізат на максимальній швидкості протягом 5 хвилин в мікрофузі.
- Соплі речовини повинні бути щільно упаковані в гранулу на дні трубки після цього кроку.
- Розчин або супернатант містить плазмідну ДНК.
- Колонка Очищення ДНК: Перенесення супернатанта в свіжу трубку з кремнезема мембранною колоною
- ДНК любить зв'язуватися зі склом в умовах високої солі.
- Біла мембрана виготовлена зі скловолокна.

- Центрифугуйте супернатант через колонку протягом 1 хвилини на максимальній швидкості в мікрофузі.
- ДНК буде пов'язана з мембраною на колоні (кремнезем)
- Промивання: Відкиньте проточний потік і помістіть колонку назад у відпрацьовану трубу. Промивна колонка з 500 мкл ПЕ. Центрифугуйте супернатант через колонку протягом 1 хвилини максимум в мікрофузі.
- ПЕ - це розчин, який допомагає змивати неспецифічно пов'язані речовини
- Відкиньте проточний і помістіть колонку назад у відпрацьовану трубу. Промивна колонка з 700 мкл ПЕ. Центрифугуйте супернатант через колонку протягом 1 хвилини максимум в мікрофузі. Відкиньте проточну і повторіть віджимання до сухої колонки.
- Помістіть колонку в свіжу пробірку центрифуги і елітуйте нуклеїнові кислоти в 50 мкл TE (pH 8.0) шляхом зв'язування протягом 1 хвилини і віджимання з максимальною швидкістю протягом 1 хвилини.
Ідентифікація плазмідної ДНК
Після виділення плазмід вони вимагають ідентифікації. Плазмідні вектори мають відомі послідовності і відображені їх основні особливості. Знання послідовності цих шматочків ДНК означає знати місця розташування місць травлення РЕ. Використовуючи Res, перетравлення плазмід до відомих розмірів допомагає у перевірці плазмідної ідентичності без необхідності повторного секвенування всієї плазміди. Поширена плазміда називається PuC18 або PuC19. «p» означає плазміду, «UC» розшифровується як Каліфорнійський університет (де він був розроблений), а 18 або 19 відносяться до різниці в MCS. Ця плазміда має 2,686 пар основ або ~ 2,7 кб (кілобаза) довжиною з єдиним сайтом eCori в MCS. Ще одна плазміда, яка цікавить вивчення молекулярної біології, називається PgLo. Ця плазміда має ген медузи в MCS, який кодує білок, який буде флуоресцентним зеленим, коли він виражається під ультрафіолетовим світлом. pgLo має довжину 5,4 кб і містить єдиний сайт eCori. Після перетравлення ферментом ці плазміди можуть бути ідентифіковані на основі поділу розмірів на агарозному гелі. Зазвичай найкраще ідентифікувати за допомогою 2 різних RE. Травлення важливо перед порівнянням розмірів, оскільки кругова ДНК мігрує через агарозу інакше, ніж лінійна ДНК. Крім того, кругова ДНК іноді може бути «супер-згорнутою» і призводити до дуже швидкої міграції, незважаючи на розмір.

Особливість карти PuC19, включаючи розташування деяких рестрикційних ферментів.

Крупним планом подання PuC19 кілька клонування сайту (MCS). Ці місця обмеження з'являються лише один раз по всій повноті плазмідної послідовності.
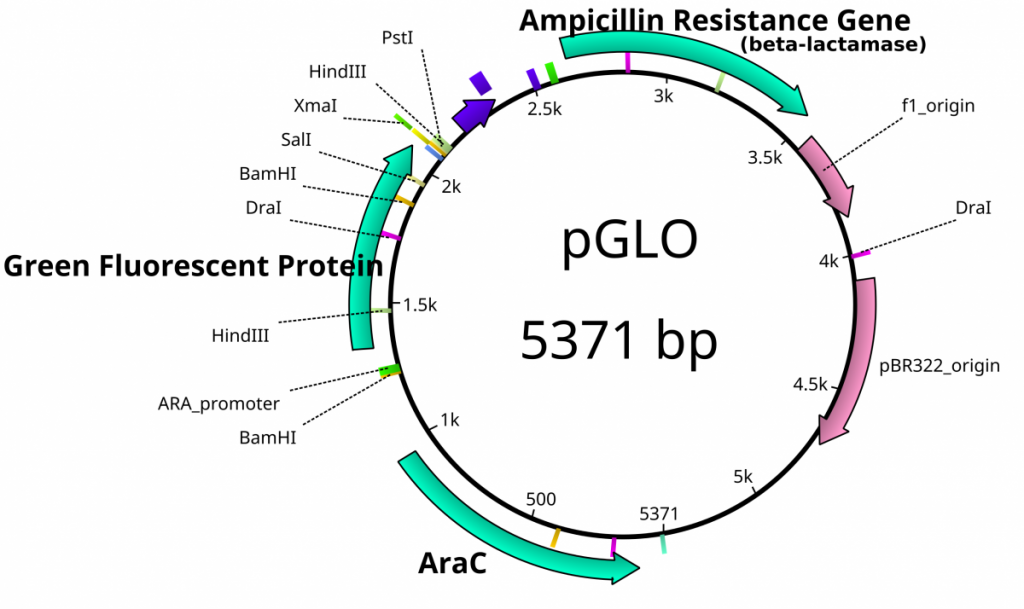
Карта плазмідних об'єктів PgLo. Це не вектор клонування, тому MCS не існує.
Вправа 2: Обмеження травлення ідентифікації плазмід
- Клас повинен готувати 2X 0,8% гелі агарози, готуючи 0.4g агарози в 50 мл TBE буфері.
- Розтопити розчин агарози в мікрохвильовці протягом 1 хвилини.
- Додайте 5 мкл безпечного розчину Sybr в розчин гелю 100 мл.
- Вилийте цей розчин в відливний лоток всередині холодильника.
- Вставте гребінець.
- До нової пробірки додають 2 мкл плазмідної ДНК до 8 мкл суміші швидкого перетравлення Eco RI.
- Буфер швидкого дайджест 1 мкл
- 1мкл швидкого дайджест Eco RI ферменту
- 6 мкл Н 2 О
- Інкубувати при 37°C протягом 10 хвилин.
- Додайте 2 мкл завантажувального буфера до суміші для травлення.
- В окремій трубці об'єднайте 3 мкл плазмідної ДНК з завантажувальним буфером 2 мкл і 7 мкл H 2 O.
- Завантажте гель з драбинкою відповідного розміру в першу смугу, завантажте перетравлену плазміду в наступні смуги, потім неперетравлену плазміду.
- 3 групи можуть навантажувати на один гель
- D = перетравлена плазміда
- U = неперетравлена плазміда
- Запустіть гель при 110 В протягом 30 хвилин і візуалізуйте на УФ-трансілюмінаторі.
- Документ за допомогою камери.
Додаткові ресурси
- Щоб отримати допомогу у вирішенні цієї проблеми, будь ласка, спробуйте активність травлення In silico.