10: Стратегії синтезу простагландинів
- Page ID
- 24282
Хімічний синтез простагландинів був свідком феноменальної активності в 1960-х і 70-х роках, в цей період органічна хімія спостерігала інтенсивний розвиток в «роз'єднанні» і «логіці» як основних інструментів синтезу. У цей період також було розроблено кілька нових реагентів для стереоселективного синтезу. Складність будови скелета ПГ поставила великий виклик для синтезу. Той факт, що молекули, що належать до цього сімейства, мали великий потенціал як кандидати на наркотики, але були доступні лише в незначних кількостях з природних джерел, був основною причиною інтенсивної активності в хімічному синтезі та скелетних модифікаціях для досліджень SAR. Система нумерації на скелеті і основні структурні особливості цього сімейства молекул можна побачити на малюнку 10.1.
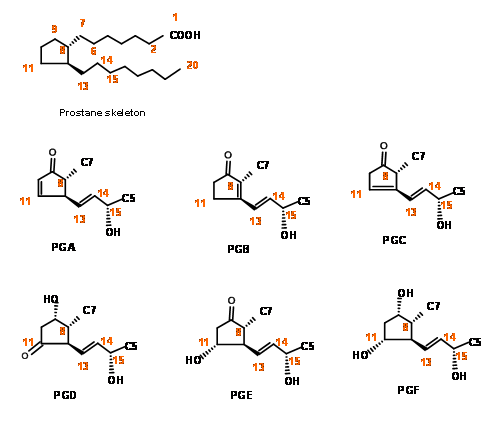
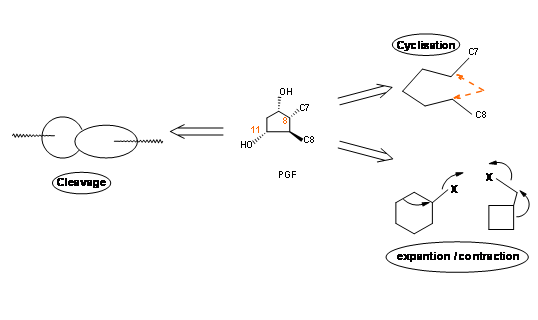
Розміщення кето-групи на C9 є делікатною операцією, оскільки такі β-гідроксикетони легко піддаються зневодненню, щоб дати скелет PGA. На п'ятичленному кільці система PGA знову може пройти готову ізомеризацію до PGB, стабільної кільцевої системи, ймовірно, через скелет PGC. При відновленні борогідридом натрію кето-група при С9 знижується до суміші 9α- і неприродних 9β- епімерів. Природний скелет PGF має конфігурацію 9α- для групи —OH. У природному скелеті PGF ми маємо чотири асиметричні центри на п'ятичленному кільці. Як ви добре знаєте, п'ятичленне кільце є конформаційно дуже гнучким. Отже, встановлення точної стереохімії на цьому кільці ставило великий виклик протягом 60-х років. може бути три основні стратегії побудови п'яти членних кільцевих систем (рис. 10.2).
- Відкрита ланцюг може бути циклічна на кільце.
- Відповідне кільце циклоалкану може бути розширене або стиснуте до п'ятичленного кільця.
- Відповідна велосипедна [l, m, n] кільцева система може бути відкрита, щоб дати п'ятичленне кільце.
Давайте розглянемо кілька видатних синтетичних зусиль, які успішно відповіли цим викликам в хімії простагландину.
Циклізація попередників відкритого ланцюга
Створення серії асиметричних центрів на відкритому ланцюзі є таким же великим завданням, як і встановлення їх на п'ятичленному кільці. Рання спроба Корі в 1968 році полягала в тому, щоб спроектувати відповідне кільце з шістьма членами, відкрити кільце до ланцюга, а потім циклізувати ланцюг regioконкретно до необхідного п'яти членів кільця (J. Am. Хім. Соц., 90, 3245 (1968)). Їх ретроаналіз і синтез показані на малюнку 10.3. Перша реакція DA встановила два найважливіших стереоцентрів, які керують рештою стереоточок на п'ятичленному кільці, утвореному реакцією альдолу. Цей синтез є хорошим прикладом для конвергентного синтезу. Вихідні матеріали надходять за двома різними маршрутами, показаними на малюнку 10.4.
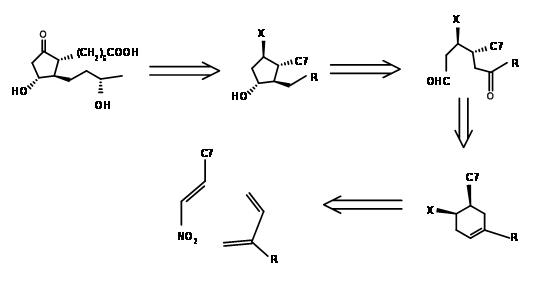
Відключення Kojima циклопентанового кільця при зв'язку C8 - C12 забезпечило відкритий ланцюг. Ретельне планування функціональних груп на запропонованому ланцюжку дозволило його групі планувати стереоцентри більш прямим способом. Їх ретроаналіз і синтетичний маршрут показані на малюнку 10.5.
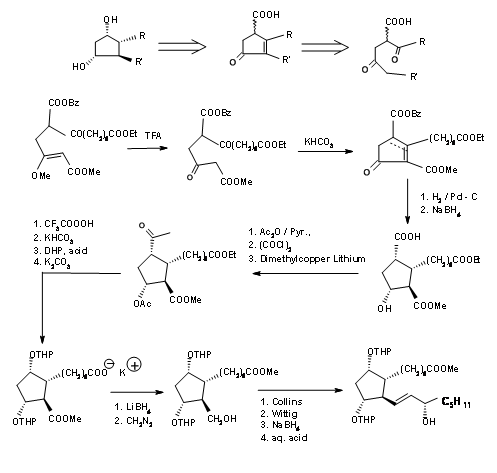
Циклопентановий кільцевий попередник
Складна проблема встановлення стереоцентрів на п'ятичленному кільці була вирішена елегантно Корі та ін. , (Тет. Летт., 311 (1970)) (рис. 10.6). Дотримуйтесь художньої точності, з якою замісники вплітаються в п'ятичленне кільце за допомогою реакцій йодолактонізації та епоксидного кільця. Приклад для майстерності в мистецтві синтезу.
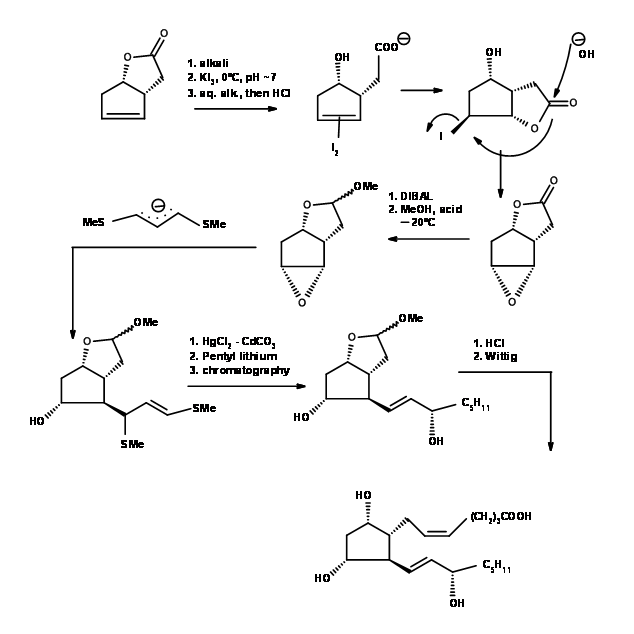
Ф.С. Альварес та ін., скористався стеричними обмеженнями через затемнення штамів у п'яти членних кільцях і плели три асиметричні центри поспіль на п'ятичленному кільці (J. Am. Хім. Соц., 94, 7823 (1972)) (рис. 10.6).
Циклогексанове кільце Попередник
Починаючи з усієї цис-циклогексан1,3,5-тріолу, школа Вудворда продемонструвала свою досконалість у мистецтві органічного синтезу. Перший крок - диференціальний захист з гліоксалевою кислотою (рис. 10.8). Відзначимо цікавий архітектурний аспект дизайну. Цей ланцюг з двох вуглецевих захисних груп врешті-решт включений в основну структуру. Зауважте, що це також виконує ще одне дуже складне завдання, а саме, розміщуючи реактивний вуглецевий ланцюг 2 в переповнену увігнуту фазу. Солволізу мезилату допомагає сусідній олефін. Зверніть увагу на елегантне планування кроку скорочення кільця.
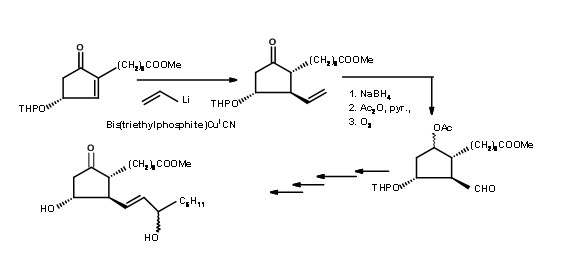
У 1973 році Корі розкрив синтез (Тет. Lett., 309 (1973)) на основі шестичленного кільця, що включає розширення кільця та скорочення кільця для досягнення мети (рис. 10.9).
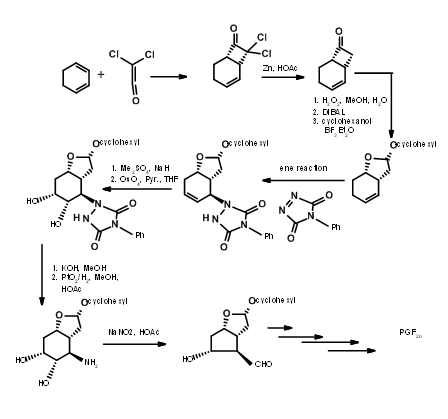
Підходи до біциклоалкану
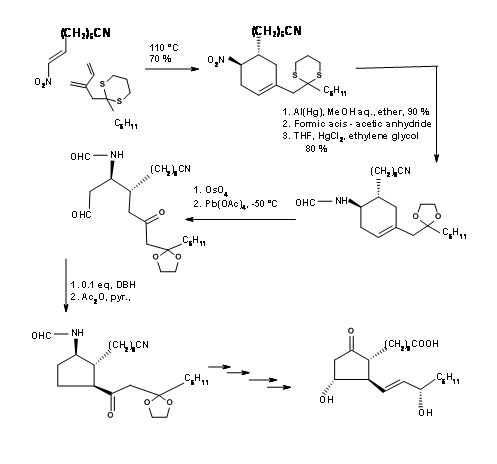
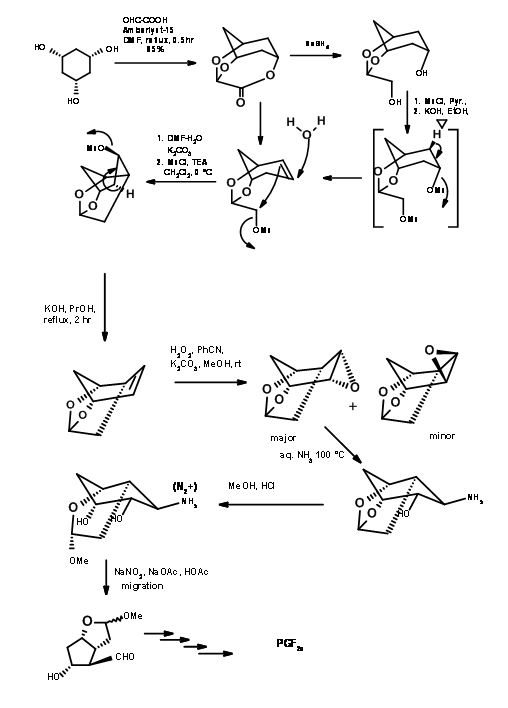
Існує кілька синтезів, заснованих на такій стратегії, щоб отримати стереохімічну перевагу кільцевої системи біциклоалкану. Тут ми обговоримо біциклогептанову стратегію Корі (J.Am. Хім. Соц., 91, 5675 (1969): Анна. Н.Ю. акад. Канд. наук, 180, 24 (1971). Цей маршрут забезпечує в'їзд всіх природних і неприродних ПГ. Він забезпечує установку для поділу енантіомерів на дуже ранній стадії за допомогою процедури амфетамінової солі на першій хіральній кислоті-спирті. Ця схема була масштабована до мультиграмм-масштабів. Ретроаналіз Корі показаний на малюнку 10.10. Цей ретроаналіз призвів до реакції DA.
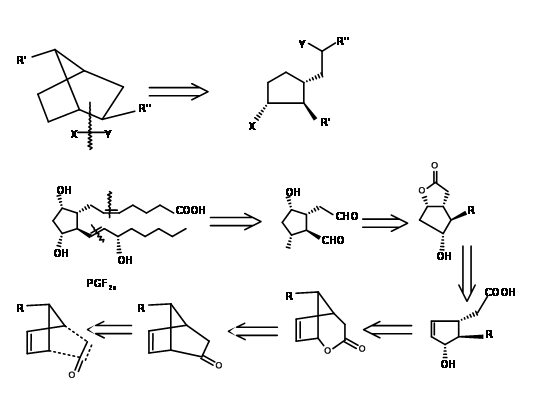
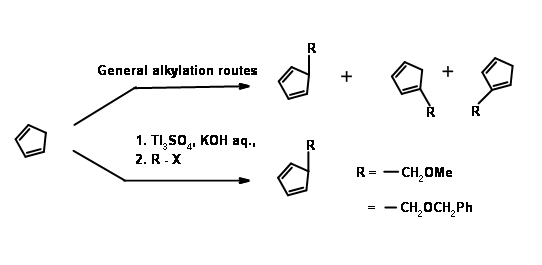
Першою проблемою був синтез циклопентадієну з алкільною групою у метиленового вуглецю. Аніонні шляхи алкілування циклопентадієну здебільшого призвели до суміші циклопентадієнів, завдяки легкій ізомеризації олефіну. Задача була вирішена алкілуванням аніоном талію (рис. 10.11). Бажаний циклопентадієн був отриманий приблизно з виходом 97%. Наступний виклик був у реакції DA.
Кетени не піддаються циклододаванню Дільса-Альдера. Вони дають циклобутанові продукти навіть з дієнами. За допомогою «маскованого попередника кетену» цю проблему було вирішено, як показано на малюнку 10.12. Утворився таким чином біцикло [2.2.1] гептан піддався окисленню Байєра-Віллігера, як очікувалося. Омилення лактону дало п'ятичленне кільце з трьома асиметричними центрами на місці. Четвертий центр був створений за допомогою реакції йодолактонізації.
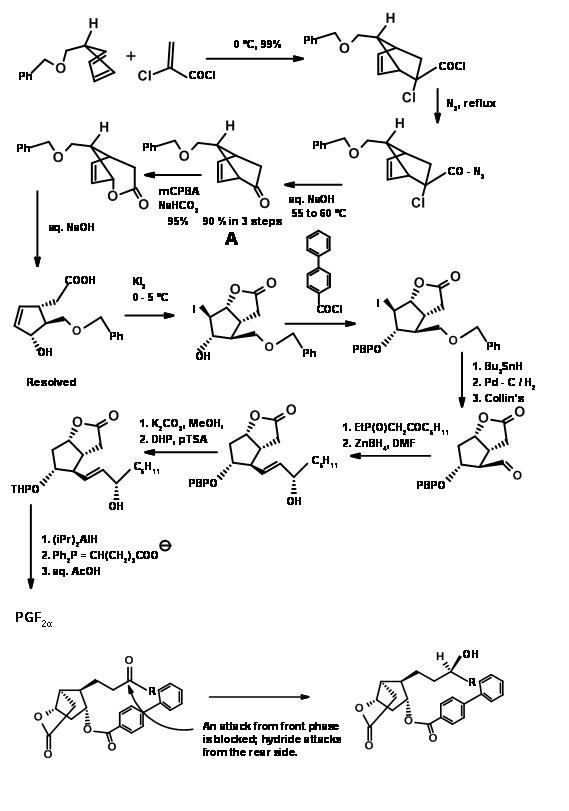
Зверніть увагу на стереохімію цього кроку йодолактонізації. Після видалення галогену —OH при C11 був захищений як п-фенілбензоїловий ефір. Цей ефір не тільки дав кристалічне похідне для очищення (корисна функція під час масштабних реакцій), але також служив захисним агентом для індукування C15- (S) - OH на більш пізній стадії. Гідрогеноліз C13 - АБО до - OH з подальшим окисленням Коллінза дав C13 альдегід, придатний для реакції Віттіга. α, β-ненасичений кетон був відновлений вибірково до C15- (S) - OH за допомогою борогідриду цинку. Ця вибірковість була приписувана екрануючим ефектом об'ємної ароїльної групи з лицьового боку, що дозволяє гідриду входити стереоселективно. Зменшення ефірової групи триалкілалюмінійгідридом дало геміацеталь для остаточного Віттіга. Зверніть увагу на диференціальні захисту, що використовуються в цьому синтезі на різних етапах.