6.3: Морфій
- Page ID
- 18684

Біосинтез алкалоїдів, отриманих бензилізохіноліну
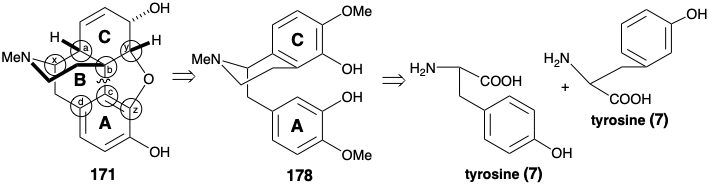
Біосинтези колхіцину і цефалотаксину включають фенілетил-ізохіноліну попередників 28 і 128. Багато алкалоїди, які отримані біологічно з двох молекул тирозину, поділяють бензил-ізохінолінові попередники загальної структури 169 (див. Нижче). Крім того, часто зберігаються обидва шість членних кілець, отриманих від арілових ядер. Обидва можуть бути ароматичними, як у берберину (170), або один може бути неароматичним, як у морфіну (171). Біогенетична стратегія берберину (170) передбачає просту дислокацію до попередника бензилізохіноліну шляхом відключення одновуглецевого електрофілу від нуклеофільного азоту та D-кільця арени. Інтактна бензилізохінолінова структура менш очевидна в звивитому багатоциклічному скелеті морфіну (171). Якщо для висококисневого C-кільця передбачається багатий електроном ароматичний попередник, то полярне роз'єднання фуранового зв'язку C-O говорить про те, що окислювальне з'єднання ароматичних A та C-кілець попередника бензилізохіноліну може генерувати B-кільце 171. Ключові проміжні продукти бензилізохіноліну 169 можуть бути отримані реакціями Манніха. Ці конденсації можуть включати утворення полярних зв'язків між фенілацетальдегідом електрофілом 173 та дофаміном (172) як бінуклеофілом.
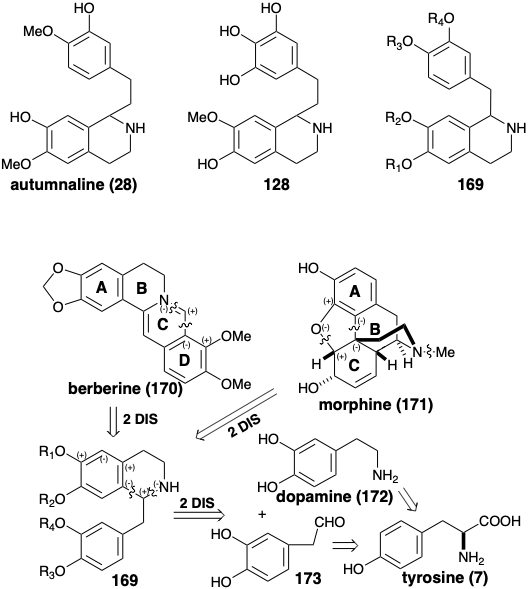
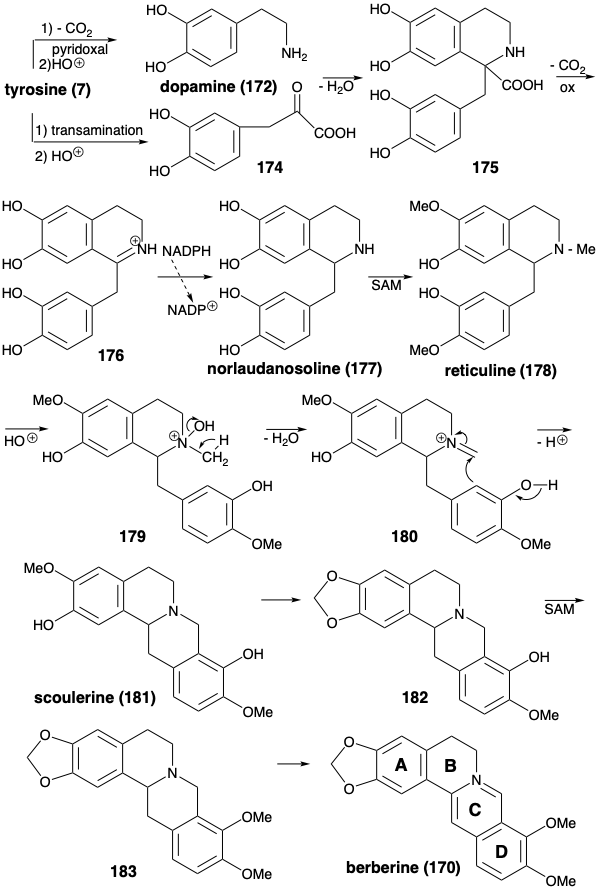
Біосинтез берберину (170) з двох молекул тирозину (див. Нижче) починається з піридоксально каталізованого декарбоксилювання та електрофільного гідроксилювання, що виробляє дофамін (172). Заміна α-аміногрупи тирозину карбонілом шляхом трансамінування і електрофільного гідроксилювання виробляє 3,4-дигідроксифенілпіровиноградну кислоту (174). Цей високореактивний кетон, а не фенілацетальдегід, служить електрофілом в реакції Манніха з дофаміном (172). Полярна конденсація високоелектрофільного карбонілу в 174 з 172 як бінуклеофіл генерує бензилізохінолінову кільцеву систему в 175. Окислювальне декарбоксилювання цієї α-амінокислоти з подальшим відновленням проміжного продукту 176 забезпечує норлауданозолін (177), з якого ретикулін (178) виробляється шляхом O та N-метилювання. N-метильна група включена в скелет берберину шляхом конденсації Манніха похідного імінію 180, отриманого зневодненням проміжного протонованого N-оксиду 179. Перетворення масиву о-метоксифенолу в продукті 181 в метилендіокси-групу в 182, метилювання і ароматизація продукту 183 забезпечує берберин (170).
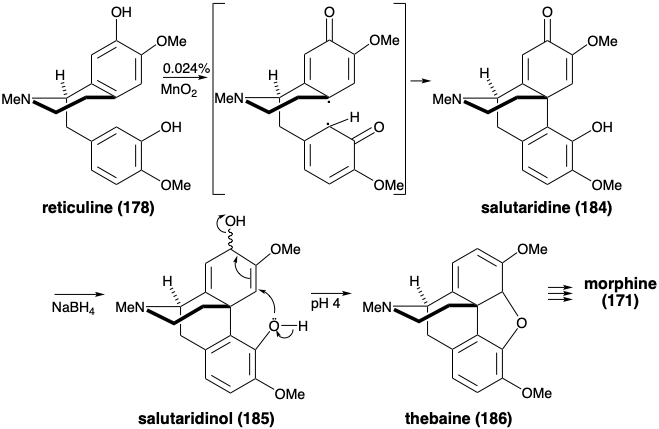
Більш топологічно складний скелет алкалоїдів морфіну також виробляється з ретикуліну (178). Таким чином, окислювальна орто-пара муфта доставляє салютаридин (184). Генерація бензофуранового кільця тебеїну (186) відбувається після регулювання рівня функціональності, що призводить до втрати функціональності з позиції 7 з 185. Цікаво, що хоча простий гідролі-сис ефіру енолу 186 може виробляти кетон 188, кисень метоксильної групи зберігається в 188. Тому повинен бути задіяний інший механізм. Можливо, деметилювання 186 відбувається через окислений проміжний продукт 187, який піддається ретроенової фрагментації. Алілова ізомеризація, відновлення та деметилювання потім доставляють морфін (171).
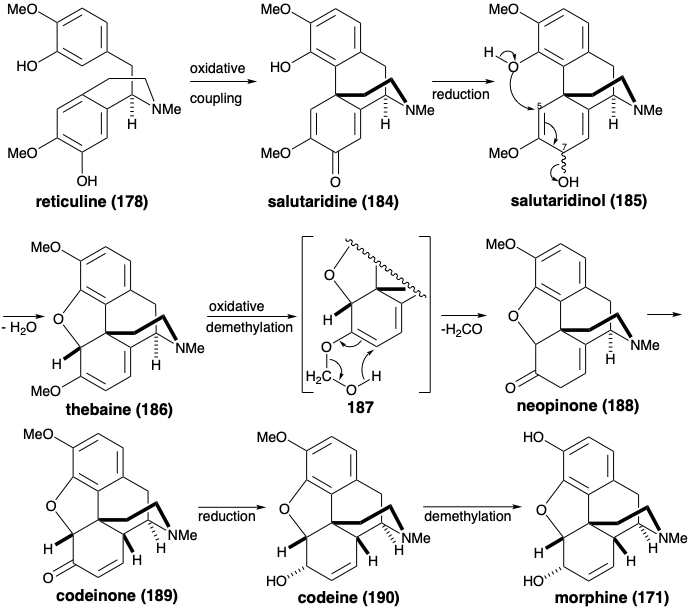
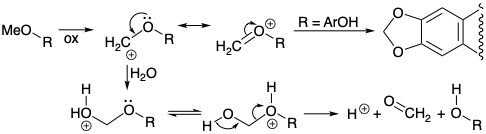
Деметилювання кодеїну (190) та ефіру енолу 187, а також перетворення фенолів орто-метокси-137 у похідні метилендіокси 139 (див. Розділ 6.2) можуть бути механістично пов'язані початковим окислювальним перетворенням метилу ефір в α-стабілізований киснем карбокатіонний проміжний продукт. Деметилювання відбуватиметься при нуклеофільному захопленні водою та фрагментації одержуваного геміацеталу формальдегіду.
Біоміметичний синтез морфіну
Морфін був зібраний в лабораторії за допомогою біоміметичної стратегії, що включає окислювальне з'єднання ретикуліну (178). 9 Окислювальне з'єднання 178 було здійснено шляхом обробки діоксидом марганцю. Салютаридин (184) був отриманий, нехай і в незначній врожайності. Зниження гідридів забезпечив алліловий спирт 185. При м'якому кислотному каталізі 185 піддався внутрішньомолекулярному S N 2' витіснення аллільного гідроксилу фенольним гідроксилом, щоб дозволити собі тебаїн (186), з якого морфін (171) може бути отриманий (vide infra).
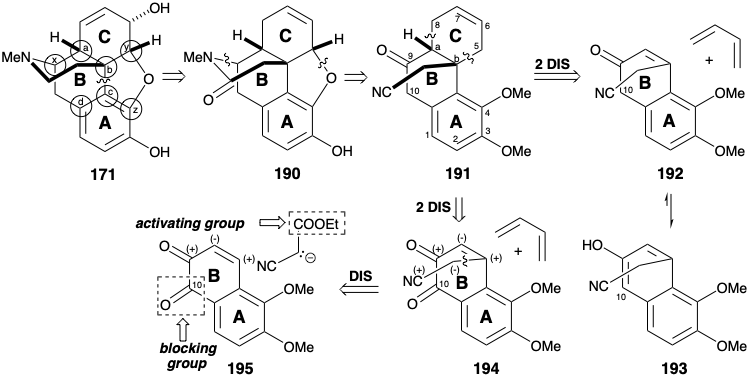
Мостовий багатоциклічний скелет морфіну має значну топологічну складність. Тому топологічний аналіз (див. Розділ 4.4) може бути корисним для синтетичного планування. Враховуючи лише карбоциклічний скелет 171, між ними існує чотири загальні атоми, a-d, і три можливі роз'єднання. З цих роз'єднань тільки одне, видалення зв'язку між загальними атомами b і c, призводить до структурного спрощення. Якщо також розглядається гетероциклічний скелет, існує також ще три загальні атоми, x, y та z Відключення зв'язків між цими останніми загальними атомами та гетероатомним кільцевим елементом, як правило, тривіальне, оскільки гетероатоми мають реактивну функціональність. Відключення b-c зв'язку (і зв'язку між загальним атомом y і киснем) передбачає такий попередник, як 178 (ретикулін), біосинтетичний прародитель алкалоїдів морфіну. Цікаво, що це єдине розщеплення зв'язку між парою загальних атомів, що призводить до спрощення морфіну вуглецевого скелета. Таким чином, розщеплення зв'язку між загальними атомами a і b призводить до проміжного з двома злитими десятьма кільцями, що було б сумнівним синтетичним викликом. Оскільки це не призводить до зниження молекулярної складності, цей вивих, ймовірно, не корисний. Розщеплення π-зв'язку між загальними атомами c і d порушує ароматичну систему і створює десятичленне кільце. Стабільність ароматичних систем зазвичай порушує синтетичні стратегії, що включають відпалювання ароматичних кілець на завершальних стадіях синтезу. Тому цей вивих теж напевно не корисний.
Стратегія Вільхи Дільса для ануляції кільця C
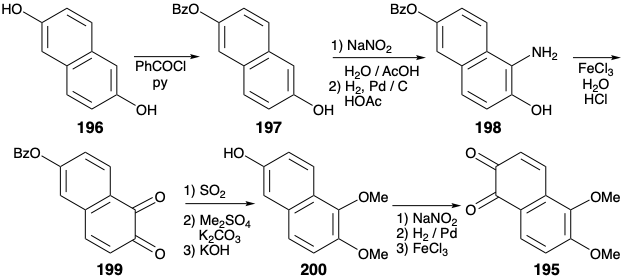
Наявність циклогексенового масиву в кільці С морфіну (171) рекомендує враховувати реакцію Дільса-Альдера для створення двох віцинальних ексендозв'язків, кожна з яких включає один загальний атом (тобто a або b) та один непоширений атом. Це топологічне спрощення було використано в першому загальному синтезі морфіну. 10 Однак зв'язок C = C в кільці C 171 знаходиться в неправильному місці. Тому з використанням тактики Дільса-Альдера як граничної умови дислокація 171 до аміду 190 створює основу для ретро-дислокації Дільса Вільхи. Щоб дозволити включення зв'язку C = C для діенофіла, 190 спочатку дислокується до 191 шляхом розщеплення вуглецево-гетероатомних зв'язків до загальних атомів x і y. карбоніл в положенні 9 в 191 забезпечує активацію для побудови Дільса-Альдера C-кільця з AB-кільця діенофіл 192 і 1,3-бутадієн, відносно багатий електронами дієн. Однак ця стратегія смертельно недосконала, оскільки очікується, що 192 буде існувати майже виключно у формі ароматичного енолу 193, що не було б реактивним діенофілом. Щоб заблокувати цю небажану енолізацію, карбонільна група може бути використана в положенні 10 (нумерація морфіну) в попереднику 194. Ціанометил бічний ланцюг може бути доповнений полярним об'єднанням нітрилостабілізованого нуклеофіла з електрофільним β вуглецем α, β-ненасиченого карбонільного масиву в 195 році. Карбоніл C-10 в 195 також сприяв би цьому Michael додавання нуклеофіла бічного ланцюга, запобігаючи енолізації енону в поєднанні з ароматизацією.
Схема, яка використовується для синтезу 195, використовує симетрію 2,6-дигідроксинафталіну (196), який легко піддається електрофільному заміщенню в α-положенні. Ефект відведення електронів бензоїльної групи в монобензоаті 197 зменшує електронну донорську здатність бензоїльованого гідроксилу. Тому нітрозування відбувається регіоспецифічно в α-положенні поруч з вільним гідроксилом. Зменшення групи нітрозо дозволяє амін 198, який окислюється до орто хінону 199. Зниження, метилювання та омилення потім доставляє фенол 200, який забезпечує 195 шляхом регіоселективного нітрозування, відновного розщеплення N-O та окислення.
Вуглецевий скелет діенофіла 194 завершується додаванням Михайла етилціаноацетатного карбоніону до 195. Після омилення, декарбоксилювання та ароматизації проміжний гідрохінон 201 окислюється, щоб дати орто хінон 194. Карбоциклічний скелет морфіну завершується циклододаванням Дільса-Альдера, яке забезпечило 191. Розробка кільця піперидину почалася зі зменшення, яке давало лактам 202 безпосередньо. Цей лактам є епімерним з морфіновим скелетом у положенні 14, імовірно, завдяки стеричному контролю підходу під час доставки водню до енолового зв'язку 9-14 C = C в 191 році.
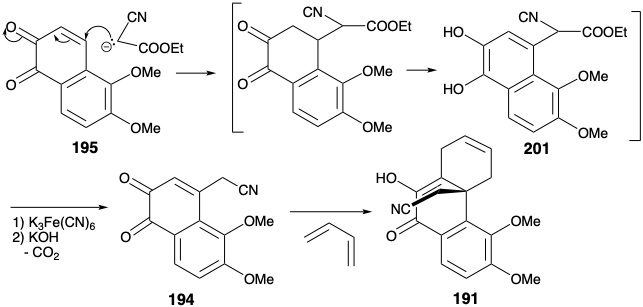
Отриманий алкоголь імовірно додає до зв'язку CN, виробляючи проміжний проміжний продукт іміноефіру, який перебудовується до лактаму 202. Примітно, що стерично набагато більш перевантажений зв'язок 9-14-C = C зменшується, тоді як зв'язок 6,7-C = C залишається невідновленим за цих умов.
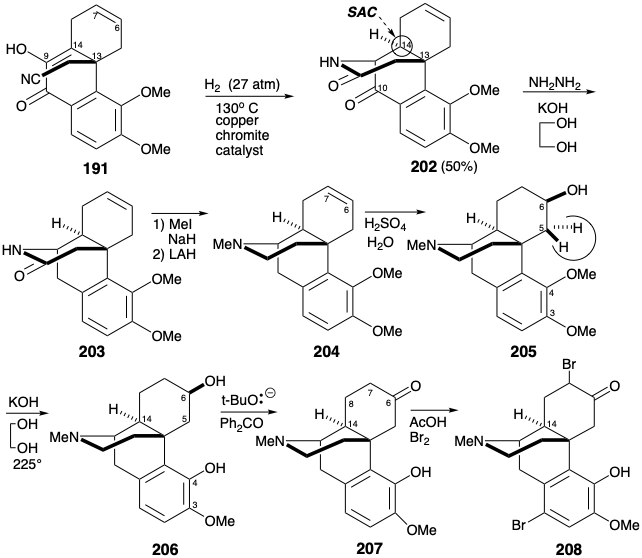
Карбоніл С-10 в 202 році, відслуживши своє призначення, потім був видалений за допомогою скорочення Вольфа-Кішнера. Після N-метилювання 203, амід карбоніл був видалений шляхом гідридного відновлення. Гідратація ізольованого π-зв'язку в 204 р. протікала повністю регіо та стереоселективно, щоб дати 205 у «врожайності до 28%». Ця вдала селективність зрозуміла з точки зору стереоелектронної переваги антиперипланарного діааксіального додавання до зв'язку C = C з додаванням нуклеофіла переважно сину до протонованого амінозамісника. Початковим наміром було деметилювати обидві ефірні групи 205 і спробувати селективне реметилювання менш стерильно перевантаженого 3-гідроксилу. Дія піридинію гідрохлориду, однак, не тільки розщеплює обидві фенольні ефірні групи, але й зневоднює вторинний спирт. На щастя, деяке деметилювання метилового ефіру C-4 спостерігалося під час перетворення 202 до 203. Це відкриття було використано шляхом розробки умов, які забезпечували 206 в 54% вихід при нагріванні з KOH в етиленгліколі. Імовірно полегшення стеричних заторів сприяє деметилюванню 4-метокси-групи шляхом зміщення фенолату гідроксидом S N 2. Завершення скелета морфіну шляхом генерації фуранового кільця вимагало значної коригування функціональності та стереохімії в 206 році. Карбоніл у положенні 6 може бути використаний як для активації 5-положення до внутрішньомолекулярної нуклеофільної атаки гідроксилом С-4, так і для забезпечення епімеризації в положенні 14. Так, окислення гідроксилу С-6 в 206 році варіацією реакції Оппенауера дало кетон 207. Для забезпечення сполучення з карбонілом С-6, необхідним для забезпечення епімеризації при C-14, був введений зв'язок C = C між вуглецями 7 і 8. Таким чином, бромування з подальшим дегідробромізацією Маттокса-Кендалла (див. Розділ 5.4) забезпечило епімеризований тозилгідразон 198.

Після гідролізу до енона 210 і відновлення до кетону 211 активація в 5-й позиції досягалася шляхом бромування двома еквівалентами брому. Незрозуміло, чому 207 не можна було перетворити безпосередньо в 212 без посередництва 210 та 211. Подальше монодегідробромізація проміжного α, α'-дибромокетону з DNPH доставлено 212. Це піддавалося циклізації в піридині з отриманням бензофурану 213 після гідролізу гідразону. Під час перетворення 207 до 208 в кільце А була введена авантативна група бромо. Це було зручно видалено під час відновлення карбонілу С-6 з гідридом алюмінію літію, щоб дати кодеїн (190), монометиловий ефір морфіну (171). Доставка гідриду до 213 відбувалася стереовибірково з більш стерильно доступною опуклої грані. Деметилювання 190 було досягнуто шляхом нуклеофільного витіснення хлоридом фенолу з протонованого ефіру.
Стратегія відпалу кільця Friedel-Crafts B
Інша стратегія синтезу морфіну (171) спрямована рішенням про генерацію В-кільця шляхом електрофільної заміщення збагаченого електроном кільцевого нуклеофіла. 11 Ця тактика вимагає тимчасової карбонільної функціональності на початковій позиції 10, яку потрібно було б видалити на заключних етапах, наприклад, шляхом зменшення попередника 214. Цільова функціональність кисню в положенні 5 може бути використана для полегшення введення залишкової функціональності C-кільця і ненасичення, а також для полегшення генерації гетероциклу азоту алкілуванням вуглецевого нуклеофіла в положенні 13. Для цього потрібна активація початкового вуглецю 15, тобто α до аміду карбонілу в 214, з нуклеофугою. Придаток амінонуклеофіла до положення 11 в попереднику 216 на 215 не може бути досягнутий полярним процесом, оскільки жодна кето група в 216 не може забезпечити електрофільну активацію в положенні 11. З іншого боку, електрофіл азоту може бути доданий до проміжного продукту, який є нуклеофільним в положенні 11 через активацію сусіднім кетоном карбонілу, наприклад, шляхом нітрозування кетону 216. ABC-кільцевий карбоциклічний скелет морфіну може бути зібраний шляхом внутрішньомолекулярної ароматичної заміщення Фріделя-Крафтса багатої електронами попередника кільця змінного струму 217. Полярний аналіз 217 рекомендує a, b-ненасичений енон електрофіл 218, який забезпечить 217 шляхом додавання карбоксиактивованого нуклеофіла.
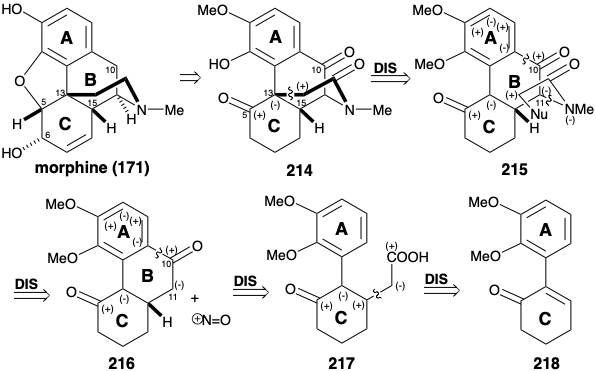
Попередник 219 з 218 може генеруватися електрофільним ароматичним підставою 220 карбонільним активованим електрофілом, 1,2-циклогександіоном. Однак стеричні затори сприяли б альтернативному регіоізомерному продукту 222. Використання альтернативного нуклеофіла, регіоселективно орто-літійованого ароматичного диетера 221 як нуклеофіла, дозволяє уникнути цієї неоднозначності. Альтернативні шляхи до 218 рекомендуються можливістю використання циклогексанону в якості більш доступного вихідного матеріалу кільця С. Таким чином, арилциклогексен 224 може бути функціоналізований послідовністю дигідроксиляції-окислення, щоб дати 218 через 219 і 223. Можливість генерації 218 безпосередньо з 224 шляхом алілового окислення страждає від неоднозначності альтернативного регіохімічного курсу, що веде до 225.
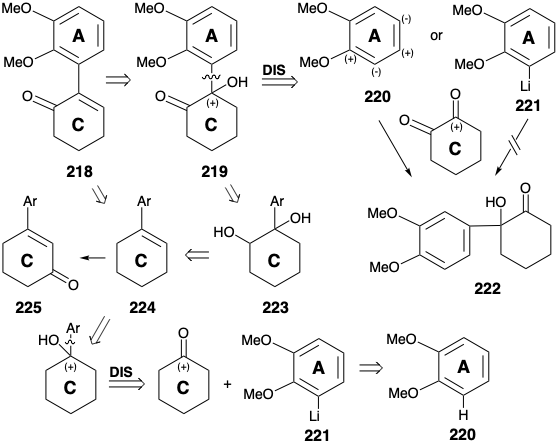
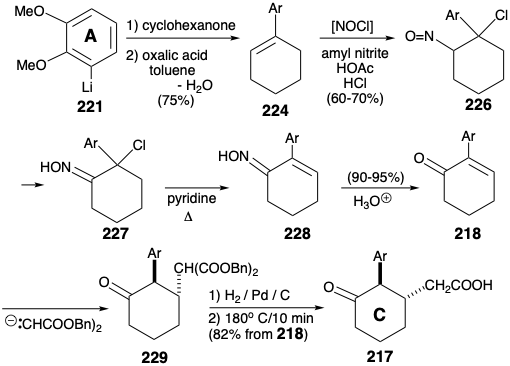
Арилциклогексен 224 легко доступний шляхом ортолітіірованія вератролу (220) з бутилітієм та реакції отриманого арилітію 221 з циклогексаноном з подальшою каталізованою кислотою дегідратацією. Бромування аллілу (з NBS) або хлорування (з т-бутилгіпохлоритом) з подальшим гідролізом та окисленням доставили необхідний енон 218. Але цей проміжний продукт був більш доступним (загальний вихід 40 - 50%) шляхом додавання нітрозилхлориду (з амілнітриту, оцтової кислоти і 30% HCl), дегідрохлорування проміжного нітрозохлориду 226 як оксиму таутомеру 227 до ненасиченого оксиму 228 і гідроліз.
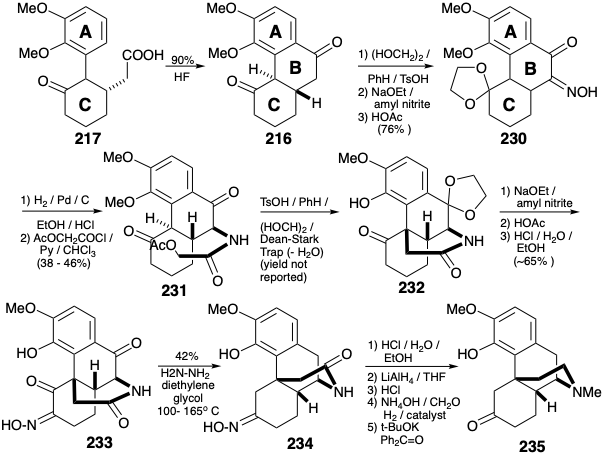
Два вуглецю, необхідні для завершення B-кільця, були додані Michael додавання дибензилмалонатного карбоніону до енону 218, гідрогенолізу отриманого 229 та декарбоксилювання. Циклізація Фріделя-Crafts 217 забезпечила B-кільце в 216 році. Диференціювання карбонілів у 216 р. може бути здійснено шляхом селективної металізації більш електрофільного карбонілу. Амінозамісник був введений шляхом нітрозування енолату. Зниження оксиму 230 в кислих умовах супроводжувалося декеталізацією. N-ацилювання доставляється α-ацетоксиацетамід 231. Внутрішньомолекулярне алкілювання та селективна кеталізація (нині менш стерильно перевантаженого карбонілу) відбулися при обробці 231 кислотою. Транспозиція карбонілу С-кільця була ініційована нітрозацією енолату 232. Декеталізація з подальшим відновленням Вольфа-Кішнера проміжного дикетоксиму 233 доставив оксим 234 видаляючи дві карбонільні групи, але не карбоніл, маскуваний оксимом. Гідроліз оксиму, відновне видалення аміду карбонілу, відновне метилювання одержуваного аміну та окислення проміжного вторинного спирту доставляють кетон 235. Перетворення аналогічного проміжного продукту 208 в морфін (171) було описано вище.
Стратегія сполученого додавання-алкілування для анулювання кільцевого кільця B
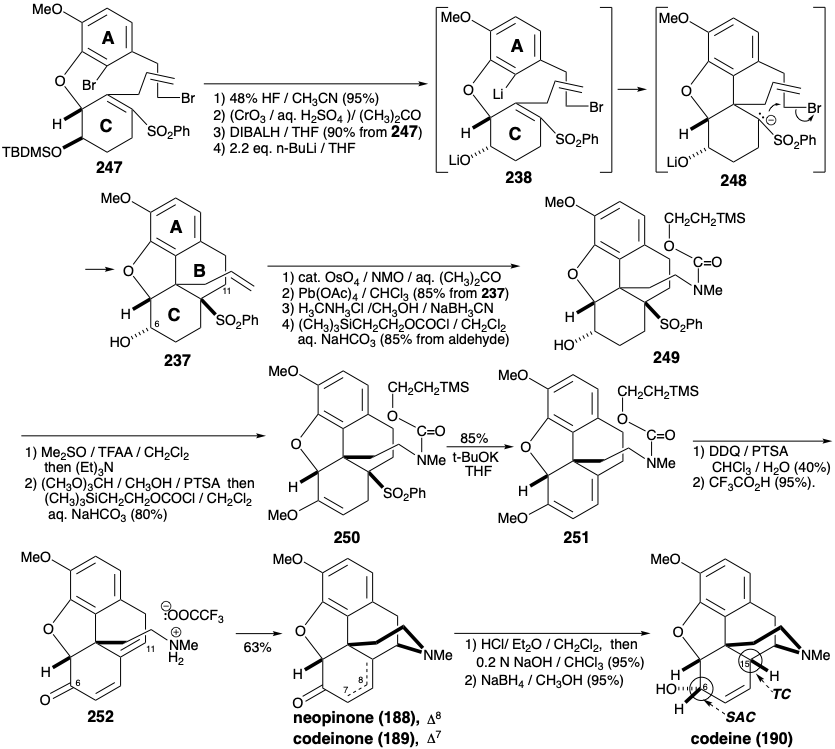
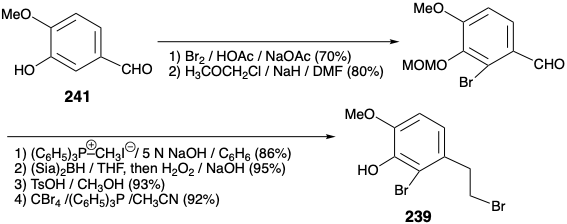
Обидва вищезгадані синтези морфіну включають: (1) велику маніпуляцію функціональною групою після побудови кілець ABC та піперидину, (2) генерацію останнього фуранового кільця та (3) залежність від карбонільних груп для активації або контролю реактивності. Зовсім інша стратегія була застосована для досягнення більш конвергентного синтезу морфіну. 12 Ця стратегія включає: (1) генерацію останнього кільця піперидину, (2) маніпуляції мінімальною функціональною групою після завершення скелетної конструкції та (3) експлуатацію сульфонільної групи для забезпечення полярної активації. Що стосується біосинтезу та попередніх синтезів морфіну, то ароматичність А-кільця рекомендує ароматичний вихідний матеріал для цього кільця. Приголосний контур між киснем С-кільця та замінниками азоту В-кільця в морфіні (171) передбачає побудову піперидинового кільця, яке використовує полярну реакційну здатність, що забезпечується цільовою функціональністю в α, β, γ, Δ-ненасичених кетонових попередниках 236. Полярне подвійне відключення В-кільця, між парою загальних атомів і між загальним і непоширеним атомом, стає можливим завдяки стратегічно розміщеній фенілсульфонілактивуючої групі в попереднику 238 з 237. Полярне відключення 238 свідчить про попередники А та С-кільця 239 та 240, які повинні бути доступні з ізованіліну (241) та симетричного 2-аллілциклогексан-1,3-діону (242) шляхом додавання функціональних груп та взаємоперетворень.
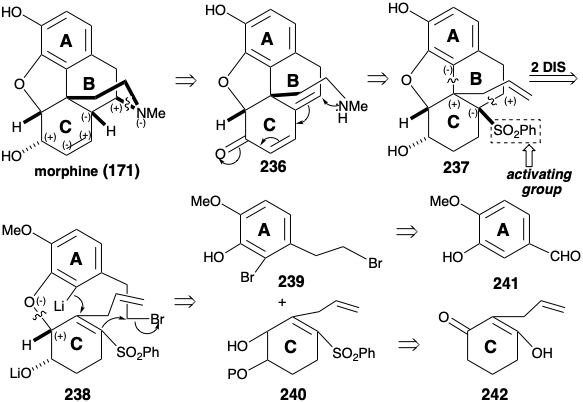
Ключ A-кільце проміжний 239 доступний у великому масштабі від ізованіліну (241) у загальному виході 40%, як зазначено нижче. Заміщення енольного гідроксилу в 2-аллілциклогексан-1,3-діон (242) на фенілсульфонільну групу здійснюється через вініллогічний ацилхлорид 243 для забезпечення 244 шляхом додавання фенілсульфінату та хлориду елімінації відповідно в 74% загального виходу. Окислювальну функціоналізацію 244 здійснювали реакцією Руботтома, тобто обробкою відповідного енол-силілового ефіру м-хлорпербензойною кислотою. Ні дефіцит електронів α, β-ненасичений сульфон, ні термінальний зв'язок C = C не окислюються в конкуренції з більш багатим електронами силіленоловим ефіром. Контроль стеричного підходу при зниженні гідридів 245 забезпечує стереоселективно алліловий спирт 246.
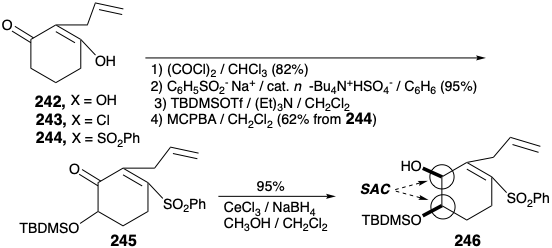
O-алкілювання 246 з 239 забезпечує ключовий проміжний 247, який зазнає чудову циклізацію при галоген-металургійному обміні. Внутрішньомолекулярне приєднання Михайла проміжного арилітію 238 відводить через сульфоновий стабілізований карбоніон 248 до 237. Побудова піпердинового кільця вимагає перетворення аллільної групи в 237 в бічну ланцюг етиламіно і кон'югації С-11 з функцією кисню в положенні 6 в 237. Окислення і етерифікація енолу забезпечує 250 з 249. Ліквідація фенілсульфінату, щоб дати 251, і гідроліз для доставки 252 встановлює етап для завершення системи морфіну кільцем шляхом генерації кільця піперидину. Таким чином, нейтралізація амонійної солі 252 генерує аміногрупу, яка піддається спонтанному внутрішньомолекулярному 1,6-Michael додавання до діенону для доставки суміші неопінону (188) і кодеінону (189) в 63% виходу. Перетворення цієї суміші через кодеїн (190) в морфін (171) було здійснено, як описано раніше Рапопортом. Необхідна конфігурація гідроксилу в положенні 6 встановлюється під час стеричного підходу контрольованої подачі гідриду до карбонільного вуглецю в 189 р. Правильна конфігурація на 15-й позиції в 190 виникає внаслідок врівноваження через загальну похідну енолу кетонів 188 і 189.