6.1: Колхіцин
- Page ID
- 18637

Біосинтетична стратегія
Використання ароматичних амінокислот, тобто фенілаланіну або тирозину, як єдиних вихідних матеріалів для колхіцину (8) вимагає підготовки семичленного С-кільця шляхом розширення ароматичного шестичленного кільця в попереднику, такому як 9. Як здійснюється розширення кільця, розглянемо пізніше. Оскільки стартові амінокислоти не мають бензилових аміногруп, цілком ймовірно, що зв'язок між кільцем С і бензиловим вуглецем утворюється шляхом електрофільного ароматичного заміщення. Оскільки початкові амінокислоти є похідними арілпропіонової кислоти, електрофіл може бути отриманий з арілпропіонового аміду, такого як 10. Зрозуміло, що зв'язок між кільцями A і C в 10 буде утворена окислювальним зв'язком багатих електронами прекурсорів арілу 11 і 12. Оскільки азот у 11, ймовірно, походить від аміногрупи в попереднику амінокислоти C-кільця, а оскільки 12 також повинен бути отриманий з а-аміноарілпропіонової кислоти, цілком ймовірно, що структура 12 повинна бути переглянута таким чином, щоб група R в 11 включала 12 як в аміді 13 (див. Нижче). Корична кислота 14, що утворюється шляхом елімінації аміаку з фенілаланіну (6), може бути прабатьком частини арилпропіонової кислоти аміду 13.
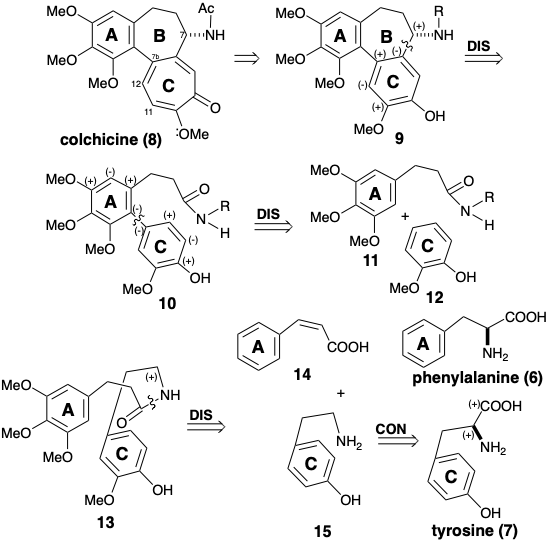
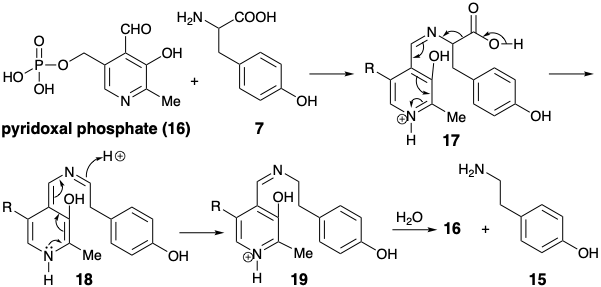
Генерація фенілетиламіну 15 шляхом декарбоксилювання тирозину (7) вимагає розщеплення зв'язку, що лежить на дисонансному контурі. Таке розщеплення досягається біосинтетично полярним процесом, який перетворює аміногрупу тимчасово в похідну, яка може стабілізувати електронний надлишок на аміновуглеці. Декарбоксилювання тирозину (7) для отримання п-гідрокси-b-фентіламіну (15) є прикладом загальної реакції амінокислот, якій сприяє кофермент піридоксальний фосфат (16). Процес ініційований утворенням шиффської бази 17. Піридиновий азот кон'югований з вуглецем а до карбоксилу і може стабілізувати електронний надлишок при цьому вуглецю. Імін азот не забезпечує полярної активації; він служить лише як сполучний атом. База Шиффа 17 легко піддається каталізованому кислотою декарбоксилювання з утворенням 18. Протонування 181 призводить до реароматизації, що доставляє базу Шиффа 19. Гідроліз доставляє фенілетиламін 15 і регенерує 16. Таким чином, піридоксальфосфат (16) діє як каталізатор, що інвертує полярну реакційну активність.
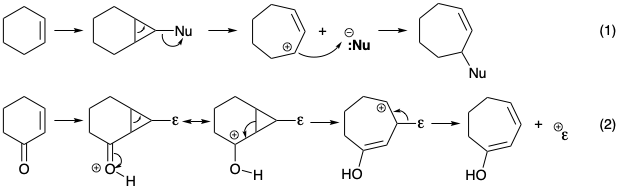
Ми зіткнулися зі стратегіями розширення полярних кілець при загальному синтезі лонгіфолену (див. Розділ 4.4). Там плавлений біциклічний проміжний продукт, породжений циклопропанацією циклогексену, піддався розщепленню злитого зв'язку спільно з відходом нуклеофуги для доставки циклогептенілового масиву (ур. 1). Аналогічний процес, що передбачає відходження електрофуги у поєднанні з розщепленням злитого зв'язку в аналогічному проміжному продукті, залежить від перестановки циклопропілкарбінілу до гомоалілкарбокатіонной перестановки (ур. 2). Наявність кисневих замінників в кільці колхіцину С говорить про можливість того, що такий механізм розширення кільця може виникнути під час біосинтетичного перетворення шестичленного попередника кільця С в семичленне кільце С.
Один вуглець фенетилового бічного ланцюга в 13, бензиловий вуглець, може бути включений в ароматичний попередник для створення семичленного кільця С. Що залишився вуглець бічної ланцюга необхідно від'єднати. Відключення цього вуглецю як карбокатіона може стабілізуватися аміногрупою як в 20. Ретросинтетичний аналіз розширення біосинтетичного кільця колхіцину, припускаючи 20 як електрофугу, передбачає циклопропілкарбініл та гомоалілкарбокацію проміжних продуктів 21 і 22. Вуглець, який вставляється в ароматичне кільце вихідного матеріалу 24, відповідає синтезу дикації 23, для якого альдегід може служити синтетичним еквівалентом.
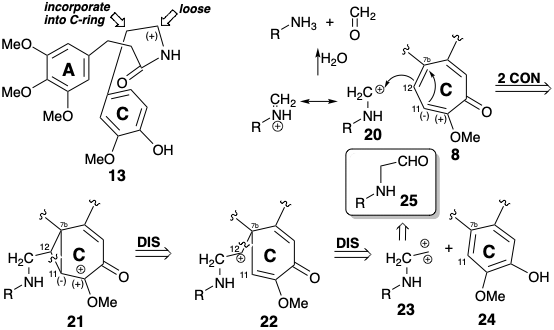
Біосинтез
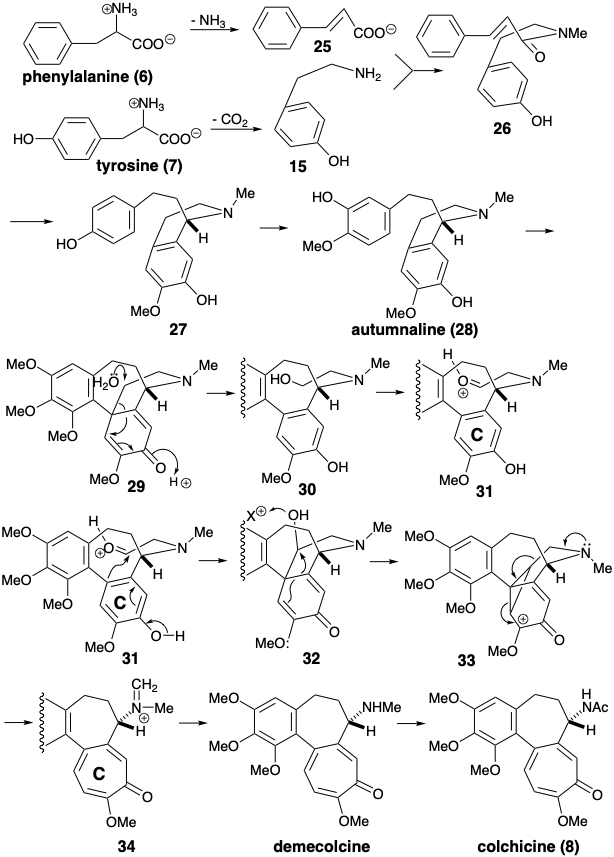
При біосинтезі колхіцин (8)\(\ce{NH3}\) виводиться з фенілаланін (6), а тирозин (7) декарбоксилюється, перед тим як утворюється амід 26 шляхом приєднання одержуваних проміжних продуктів 25 і 15. Семичленне кільце B створюється енантіоселективним внутрішньомолекулярним електрофільним ароматичним заміщенням, що дає 27 і окислювальну зв'язок 28, яка забезпечує 29. Функціоналізація очевидно неактивованого метилену в 29 здійснюється послідовністю, що включає полярну гідролітичну фрагментацію до 30 з подальшим окисленням до альдегіду, придатного для введення в шестичленний попередник кільця С в 31. Розширення арілового кільця для генерації семичленного трополону ініціюється внутрішньомолекулярним електрофільним ароматичним заміщенням. Внутрішньомолекулярне алкілювання 32 з подальшою фрагментацією проміжного циклопропану 33 виробляє кільцевий розширений скелет у 34 біосинтетичної мішені. Остаточний гідроліз групи іммінію, N-деметилювання і N-ацетилювання забезпечує колхіцин (8).
Молекулярні характеристики
Стабільність ароматичних похідних часто використовується в синтезі стратегіями, які включають попередньо сформовані ароматичні фрагменти. Так, в біосинтезі колхіцину (8) ароматичне А-кільце отримують з попередньо сформованого ароматичного кільця фенілаланіна (6). Чотири різні загальні синтези колхіцину, які будуть розглянуті в цьому розділі, приймають цю саму стратегію. Однак, на відміну від біосинтезу, загальний синтез використовує повністю функціоналізовані ароматичні вихідні матеріали. Це пов'язано з тим, що регіоселективні гідроксилювання, які досягаються ферментативно в біосинтезі, не так легко досягаються в лабораторії.
Також слід зазначити, що кільце С колхіцину (8) містить дві функціональні групи, що забезпечують електрофільну активацію на сусідніх атомах вуглецю, дисонанс полярної реактивності. Таким чином, ці функціональні групи не можуть бути використані безпосередньо в полярній реакції (тобто без umpo′lung) для створення C-C-зв'язку дисонансного контуру між цими атомами вуглецю. У кожному з наступних синтезів семичленне кільце C додається до попередника AB-кільця. У кожному випадку для відпалу кільця С використовується різна стратегія.
Ключові стратегії, спрямовані на проміжні продукти для Conchicine
Два синтези колхіцину використовували легкодоступний ключовий проміжний продукт, пурпурогаллін (34), для кільцевого фрагмента AB 8 і утворили семичленне C-кільце неполярними реакціями. Аналіз полярної реактивності 34 передбачає синтез ароматичного попередника 36. Таким чином, придаток до 36 В-кільця в 34 може бути полегшений додаванням активуючого карбоксилу, як у 35, який може генеруватися з 36 і 37 двома полярними реакціями, що утворюють зв'язок. Насправді ароматичний вихідний матеріал 36 також може бути попередником тимчасово-мостового синтетичного еквівалента 39 (vide infra) синтону 37.
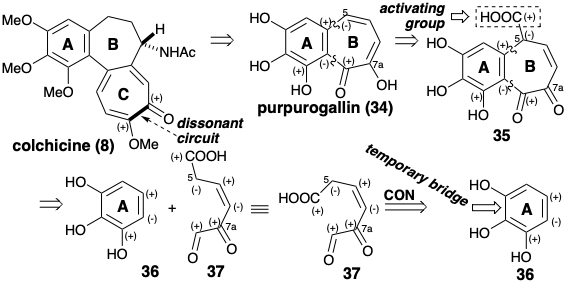
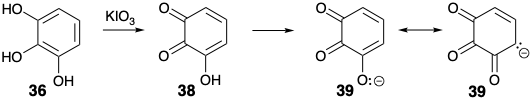
Пурпурогаллін (34) був відомим продуктом від окислення пірогаллолу (36). Ймовірно, він утворюється димеризацією 3-гідрокси-о-хінону (38). Початкове додавання Майкла енолату 39 до 38 дати 40 з подальшою внутрішньомолекулярною конденсацією альдолу призводить до трициклічного проміжного продукту 41 (див. Нижче). Це розщеплюється в ретро-реакції Дікмана, характерній для β-дикетонів, для отримання біциклічної карбонової кислоти 37, яку можна виділити. Ця бордова β-кето-кислота легко піддається декарбоксилювання, щоб доставити 28.
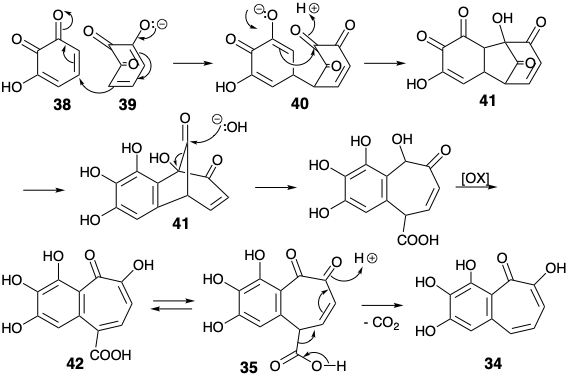
Обидва синтези, що використовують 34 для кілець A та B, спростили ціль, нехтуючи групою ацетамідо. Це може бути введено бензиловим окисленням після завершення вуглецевого скелета мішені 8. Обидва синтези будують спрощену мішень 43 з похідної бензосуберону 44, в якій карбонільна функціональна група забезпечує активацію для відпалу кільця С. Ешенмошер 1 готують 39а шляхом зменшення триметилового ефіру 45 пурпурогалліну (34) через 46, 47 і 48.
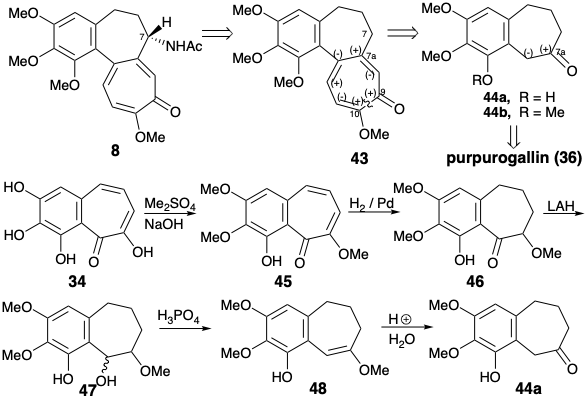
Стратегія циклоаддиції-перициклічної перестановки для кільця C
Стратегія Ешенмошера 1 для відпалу семичленного кільця C полягала в тому, щоб побудувати дієну на 44a, потім побудувати шестичленний карбоцикл за допомогою реакції Дільса-Альдера діена і, нарешті, розширити шість до семи членних кільце за допомогою перициклічної перестановки норкарадіена. Легка взаємоконверсія циклогептатрієнів, таких як 49, з норкарадіенами, такими як 50, є відомою [3.3] сигматропною (Cope) перебудовою, яка спричинена на користь циклогептатрієнів шляхом полегшення кільцевого деформації, пов'язаного з розщепленням циклопропану. Сумнівно, що стратегія була продумана шляхом суворого ретросинтетичного аналізу, оскільки перетворення 49 на 43, безумовно, вимагатиме великих маніпуляцій функціональної групи. Рішення про використання 49 в якості попередника для 43 майже напевно розвинулося як наслідок рішень про використання: (1) a Cope перестановка norcaradiene.
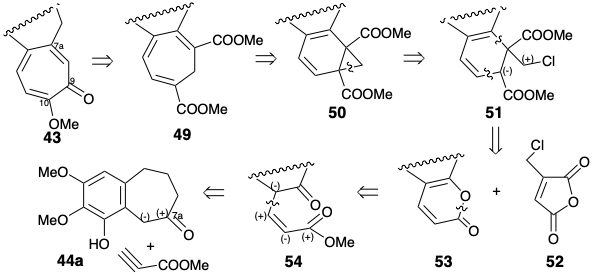
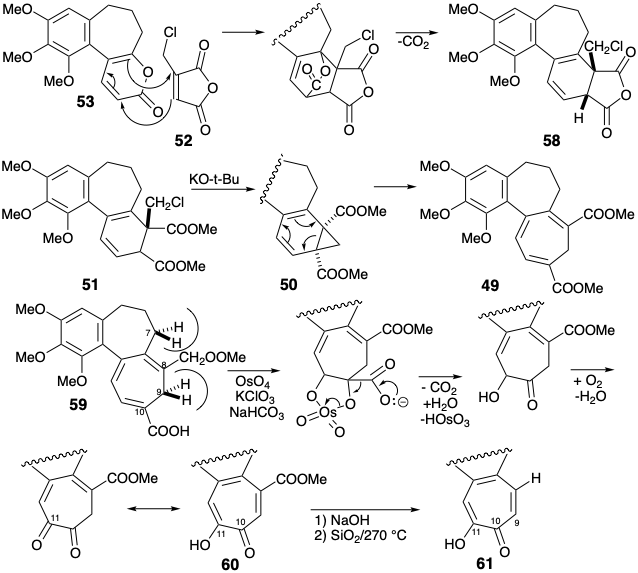
Тому хлорметилмалеїновий ангідрид (52) ідеально підходить для циклоадди до відносно багатого електронами дієну 53. Можливо, більш очевидним вивихом 50 було б 53, а циклопропен 55. Ця гілка ретросинтетичного дерева, швидше за все, розглядатиметься першою, оскільки вона забезпечить більш конвергентний синтез. Таким чином, реакція 55 з α-піроном 53 доставляла б 50 безпосередньо циклододаванням вільхи Дільса, а потім in situ ретро Diels Alder циклоелімінація вуглекислого газу з проміжного 56.
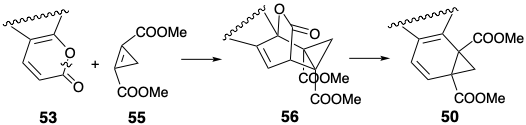
Альтернативний попередник, діенофіл 52 з хлорометилзамісником, пропонується шляхом полярного відключення від 50 до 51, що може бути отримано з 53 послідовністю Дільса-Альдера-ретро Дільса-Альдера. Діенофіл 52, а не 55 був обраний, оскільки він більш доступний, ніж 55. Додавання Дільса-Альдера від 52 до 53 з подальшим ретро-ліквідацією Дільса-Альдера\(\ce{CO2}\) з проміжного аддукту, процесу обміну карбоніл-алкенів, забезпечить циклогексадієн 51. Рушійною силою процесу є генерація відносно стабільного зв'язку C = O в обмін на зв'язок C = C діенофіла 52. Діен 53 - це енол-лактон, що виводиться з кислоти 54. Полярний аналіз 54 передбачає побудову з 44a та метилпропіолату полярним 1,4-додаванням. Очевидне алкілювання Майкла 44а метилпропіолатом забезпечується піроном 57. Хоча 44a міг дати 57 через пряме доповнення Майкла до інеона, насправді реакція була більш складною. Вона передбачала участь фенолатного аніона. Таким чином, електрофіл доставлявся внутрішньомолекулярно до досить перешкодженим бензиловим вуглецю 44а.
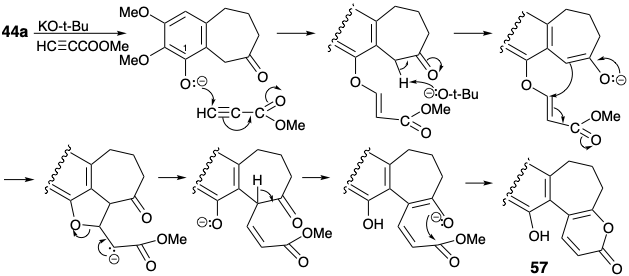
Після метилювання фенолу 57 відпал циклогексадієну 58 був досягнутий за допомогою добре відомої реакції циклоаддиції-циклоелімінації α-піронів. Базово-каталізоване внутрішньомолекулярне алкілювання діефіру 51 з 58 призвело, через норкарадієн 50, до циклогептатріену 49. Найменш перешкодженим ефіру в 49 був легко гідролізований вибірково, і результуюча кислота дозволила трополон 61 через осмій каталізований віцинальний гідроксиляції-декарбоксилювання, омилення залишився ефіру в 60 і другий декарбоксилювання.
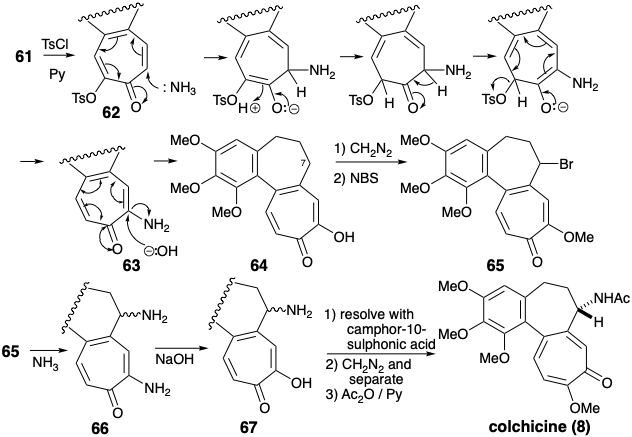
На жаль, послідовність призводить до кисневої функції на C-11, а не C-9, як це потрібно для колхіцину. Транспозиція функціональної групи була досягнута за допомогою добре прецедентної послідовності нуклеофільних переміщень на похідній тозилату 62 з 61 спочатку з\(\ce{NH3}\) дати 63, потім з -OH доставити 64. Потім трополон 64 був метильований і функціоналізований при С-7 шляхом аллілільного бромування з N-бромсукцинімідом для забезпечення 65. Нуклеофільне витіснення броміду аміаком дало необхідний С-7 амін, що супроводжується аммонолізом трополону - вінологічного ефіру. Омилення отриманого вінілогічного аміду 66 дало 67, що дозволило колхіцин при метилюванні та ацетилюванні.
Стратегія ацилоїну для кільця C
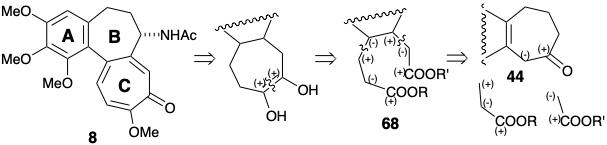
Стратегія Ван Тамелена для відпалювання С-кільця з віцинальною кисневою функціональністю визнає застосовність внутрішньомолекулярної ацилоїнової реакції для створення дисонантного контуру між віцинальними електрофільними активуючими групами. 2 Подальший полярний аналіз передбачає синтез необхідного проміжного діефіру 68 шляхом використання полярної активації, що забезпечується карбонільною групою в попереднику AB-кільця 44. Таким чином, придаток оцтової та пропіонової кислоти бічних ланцюгів повинен бути здійснений відповідно реакцією Реформатського та алкілуванням Михайла.
Пара ізомерних гідроксидікислот 69c і 69t була отримана Михайлівським алкілуванням 44b акрилонітрилом, Реформатської реакцією проміжного кетонітрилу і гідролізом. Гідроксильна група маскувалася внутрішньомолекулярно під лактон, а решта карбоксильна група була метильована. Лише один з ізомерних ефірів лактону 70 пройшов ацилоїнову реакцію, яка забезпечувала 71. На жаль, це був незначний ізомер 70t, з осьовим карбомет-оксиметилзамісником. Ефірні групи в основному ізомері 70c не могли легко досягти зіставлення, придатного для внутрішньомолекулярної реакції ацилоїну. Продукт ацилоїну 71 окислювався до 72 з Cu (II) і додатково окислювався NBS, щоб забезпечити трополон 64. Метилювання та бромування доставили бромід 65, проміжний продукт, який також готував Ешенмошер. Заміна аміногрупи на бромозамісник в 65 з подальшим гідролізом, реметилуванням вінілової карбонової кислоти, а N-ацетилювання виробляють колхіцин 8.
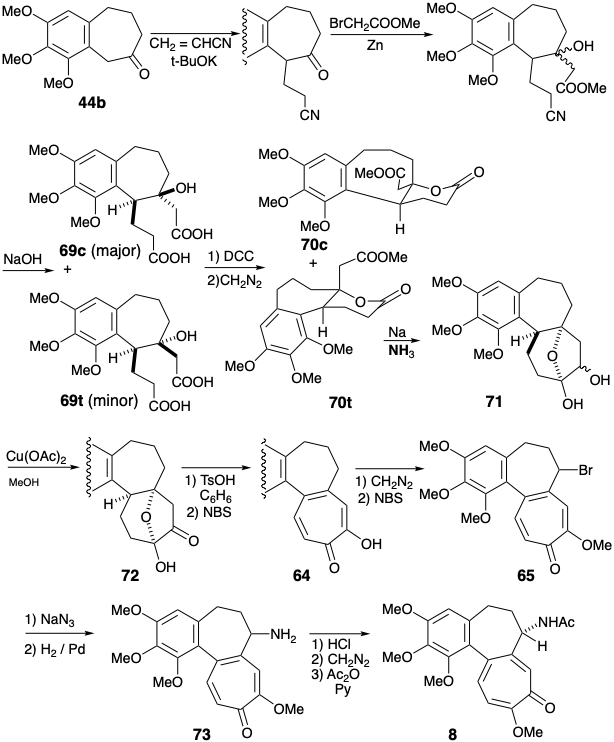
Обширні маніпуляції функціональними групами після завершення вуглецевого скелета були потрібні в синтезі Ешенмошера, оскільки була прийнята стратегія відпалу для кільця С, яка ігнорувала функціональність, пов'язану з ціллю. Ван Тамелен міг завершити свій синтез менш функціональними груповими маніпуляціями, оскільки більше цільової функціональності, яка була використана для полегшення побудови скелета, була присутня після завершення C-кільця.
Стратегія формування полярних облігацій, спричиненої цільовою функціональністю
Інший синтез колхіцину (8), який також включає відпалювання кільця С на попередньо сформованому проміжному кільці AB-кільця, був розроблений Р.Б. Вудвордом. 3 Стратегія є унікальною у включенні 7-амінозамінника на ранньому етапі та в широкому використанні цільової функціональності, пов'язаної з метою полегшення будівництва скелета вуглецю. Вивих мішені 8 до похідної 74, в якій 8-положення також блокується, дозволяє селективно вводити кисень при С-10. У синтетичному еквіваленті 75 синтону 74 ароматичне кільце ізотіазолу маскує як амінозамінник, так і С-8. Відпал C-кільця може бути досягнутий циклізацією Дікмана 77 → 76, що використовує полярну активацію, забезпечену карбонілом C-9 та активуючою карбометоксильною групою, доданою до С-10 у попереднику 77. Полярна активація, що забезпечується цією карбометильною групою в 77, передбачає електрофільну ароматичну заміну 78 → 77 для відпалу В-кільця. Звичайно, для вираження відповідної електрофільності карбометоксильна група в 77 повинна бути кон'югована з γ-положенням, як у 78. Ізотіазол також служить тимчасовим мостом у 78, що допомагає ентропічно в циклізації 78 → 77. Карбометоксил у 78 також дозволяє полярну розробку масиву дієнового ефіру з попередника ізотіазолу альдегіду 79 шляхом стабілізації карбоніону в фрагменті іліду 80. Атом сірки в ізотіазольному кільці навіть забезпечує активацію для генерації карбоніону а до сірки в 78, що дозволяє полярне з'єднання карбометоксилу, необхідного в 77. Широке стратегічне використання одиниці ізотіазолу в стратегії Вудворда є відмінною рисою цього синтетичного плану.

Після циклізації Дікмана 81 монокетон 75 С-9 селективно окислюється на С-10 полярною реакцією, що використовує нуклеофільну активацію при С-10, що забезпечується карбонільною групою С-9. Нуклеофільна атака карбоніоном С-10 на сірчаний електрофіл призводить до окислення С-10 (і супутнього відновлення сірки). Отриманий тіокетал 82 гідролізується до 83, що дозволяє продукт ацетилювання енолу 84. Енодиолат, отриманий шляхом омилення діацетату 84 легко окислюється, щоб доставити 85. Десульфуризація 85 за допомогою нікелю Рейні видаляє ізотіазольну маскувальну групу. Зменшення отриманого іміну 86 з подальшим ацетилюванням забезпечує 87, тобто N-метил доставити колхіцин (8).
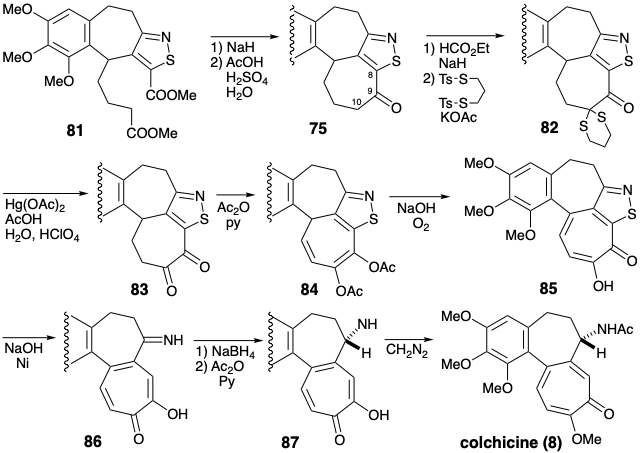
Синтез Вудворда ключового проміжного 81 зосереджений навколо нового ароматичного ізотіазольного кільця. Вихідний матеріал, ізотіазол 88, легко доступний з метилу β-амінокротонату, енаміну, отриманого з метилацетоацетату. Кон'югована метильна група в 88 легко бромується з NBS. Алкілування\(\ce{Ph3P}\) дозволяє фосфонію бромід 89, що дає ілід 90 при депротонації. Віттіг олефінація 3,4,5-триметоксибензальдегіду (91) з лідом 90 виробляє алкен 92, який вибірково гідрогенізований димідом. Каталітичне гідрування 92 було виключено сприйнятливістю ізотіазольного кільця до гідрогенолізу. Гідридне відновлення ефіру 93 і часткове окислення проміжного кон'югованого карбінолу дали альдегід 94. Віттіг олефінація 94 з ілідом 95 з подальшим омиленням і цис-транс-ізомеризацією забезпечується 96.
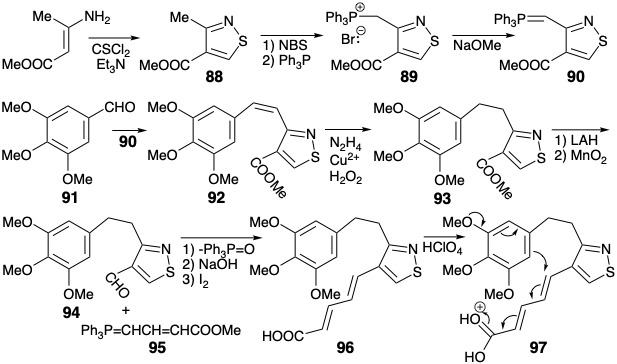
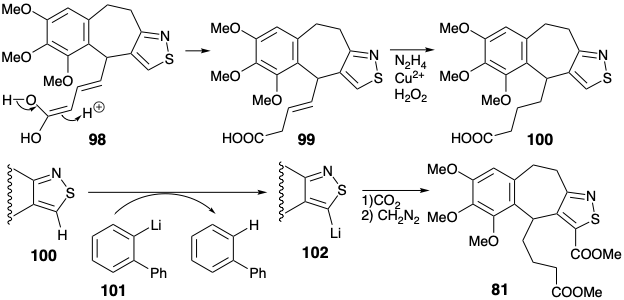
Цей дієн піддався внутрішньомолекулярному електрофільному ароматичному заміщенню після обробки соляною кислотою. Тимчасовий міст, забезпечений ізотіазольним кільцем, безсумнівно, полегшує цю циклізацію, сприятливо зіставляючи реагуючі вуглецеві центри. Селективне відновлення продукту циклізації 99 з диимидом дало 100. Кінцевий вуглець, необхідний для кільця С, був введений шляхом карбонілювання похідного органолітію 102 з тіазолу 100. Таким чином, селективне металізація в присутності карбоксилату була досягнута при відносно ненуклеофільному, стерильно обтяженому аріл-літієм 101. Літійований тіазол 102 дав ключовий проміжний продукт 81 після карбонізації з подальшим метилуванням.
Гіпотетично біоміметична стратегія
Четвертий синтез колхіцину, розроблений Скоттом 4, відрізняється від попередніх трьох своєю стратегією побудови скелета. Скотт зібрав проміжний продукт, що містить кільця A і C, а потім створив кільце B за допомогою внутрішньомолекулярної окислювальної зв'язку. Ця стратегія була заснована на гіпотетичному механізмі біосинтезу колхіцину, який зараз відомо, що не працює. У цьому механізмі, на відміну від фактичного біосинтетичного механізму (див. Вище), генерація трополонового С-кільця шляхом кільцевого розширення ароматичного попередника передує окислювальному зв'язку, яке створює В-кільце.
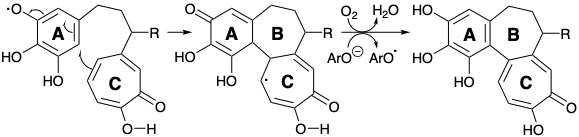
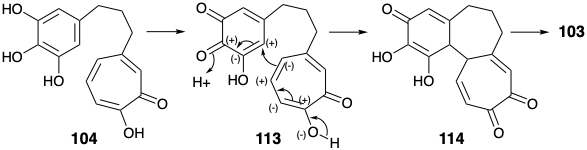
Тактика введення аміногрупи колхіцину в кінці синтезу була добре прецедентною. Таким чином, стратегія Скотта починається з дислокації цілі 8 до попередника 103. Зв'язок між ариловими і трополоновими кільцями лежить по дисонансному контуру між функціональністю кисню в положеннях 1 + 9, 1 + 10, 3 + 9 або 3 + 10. Таким чином, полярний аналіз показує, що формування цього зв'язку не може бути досягнуто полярним процесом з використанням цих функціональних груп для забезпечення активації, оскільки потрібно об'єднання двох нуклеофільних центрів. Такий процес може бути досягнутий окислювальним шляхом, припускаючи дислокацію 103 до попередника 104 з двома моноцикліками, з'єднаними простим триметиленовим містком. Полярна реакційна здатність, що забезпечується карбонільною групою в 104, може бути посилена шляхом активації карбоксильних груп у попереднику 105, що дозволяє формувати полярний зв'язок між електрофільним проміжним продуктом 106 та нуклеофілом, отриманим від 107 шляхом депротонації. Неочевидний вибір 107 як попередника для 104, безсумнівно, був продиктований його готовністю з пурпурогалліну 34 шляхом селективного окислювального розщеплення відносно багатого електронами арилового кільця.
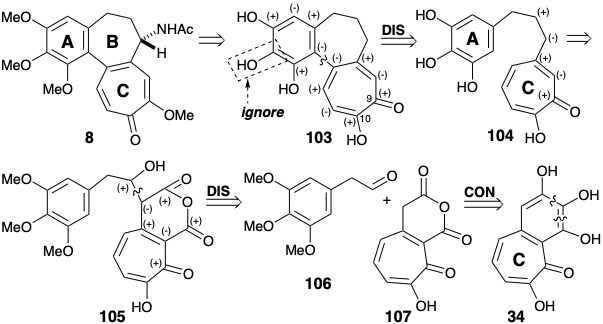
Так, окислення 34 пероксидом водню з подальшим зневодненням дало енол ангідрид 107. Цікаво, що Скотт використовував пурпурогаллін (34) як попередник для C-кільця колхіцину на відміну від Ешенмошера та Ван Тамелена, які побудували кільця A і B колхіцину з 34.
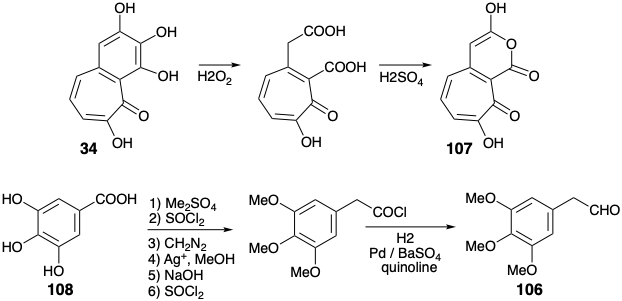
Синтон А-кільця 3,4,5-триметоксиацетальдегід (106) був отриманий шляхом омологації Арндта-Ейстерта 3,4,5-тригідроксибензойної кислоти (108). Об'єднання електрофільного кільцевого синтону 106 з нуклеофільним С-кільцевим синтоном 107 найкраще було досягнуто термічно без базового каталізу шляхом процесу, який починається з альдольної конденсації. При 100° C утворювався лактон 110, імовірно, шляхом декарбоксилювання проміжної бордової β-кето-кислоти 109. Подальше нагрівання 110 при 190° C дало 112, імовірно, шляхом декарбоксилювання проміжної β-кето-кислоти 111. Зменшення і деметилювання дали кільцю A+C проміжний 104. Збагачений електронами продукт 103 з бажаного окислювального зв'язку 104 був дуже сприйнятливий до небажаного подальшого окислення. Тим не менш, м'який окислювач\(\ce{FeCl3}\), в двофазній системі дав 103 після паперової хроматографії в інертній атмосфері, хоча і з низьким виходом (5%).
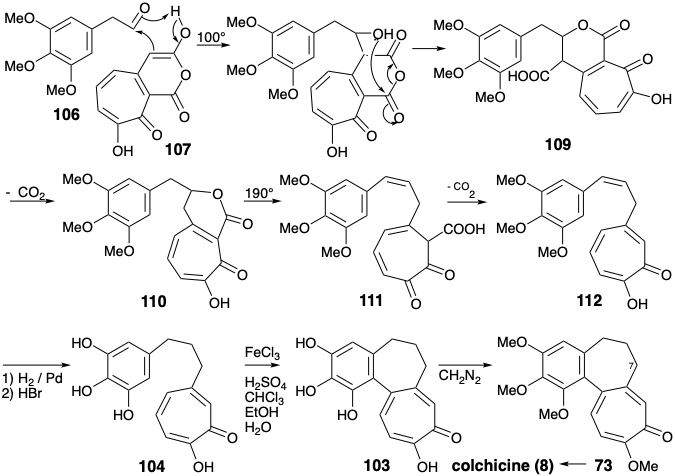
Було висловлено припущення, що відпалювання може включати іонний процес, а не радикальне з'єднання, яке спочатку передбачалося. 5 Таким чином, використовуючи функціональність арилового кисню в положенні 2, яка була проігнорована в полярному аналізі 103 вище, видно, що зв'язок між кільцями арила та трополону лежить на приголосному контурі між позиціями 2 і 10. Це дозволяє Майклу додавати нуклеофіла трополону до енонового електрофілу, як показано в 113, доставити 114. Остаточне коригування функціоналу дало 8 від 103 до 73, як обговорювалося раніше.