6.2: Цефалотаксин
- Page ID
- 18652

Біосинтез цефалотаксину (115) передбачає конвергентну стратегію, яка збирає складний багатоциклічний скелет з двох попередників ароматичних амінокислот, фенілаланіну (6) і тирозину (7). Як і в біосинтезі колхіцину (8), одне ароматичне кільце включено неушкодженим, а інше широко модифіковано. Таким чином, в біосинтезі колхіцину (8) семичленне С-кільце розробляється шляхом одновуглецевого розширення ароматичного кільця, отриманого тирозину. На відміну від цього, біосинтетична стратегія цефалотаксину (115) використовує одновуглецеве кільце скорочення для отримання п'ятичленного С-кільця з шестичленного кільця, похідного фенілаланіну. Логіка стратегії базується на: (1) готовності високооксигенізованих похідних циклогексилу, таких як 119 шляхом окислювального метаболізму ароматичних попередників та (2) можливості екструдування атома вуглецю у вигляді вуглекислого газу з α-дикетону шляхом перестановки бензилової кислоти на α- гідроксикислота. Це говорить про α-гідроксикислоту 116 як попередник 115.
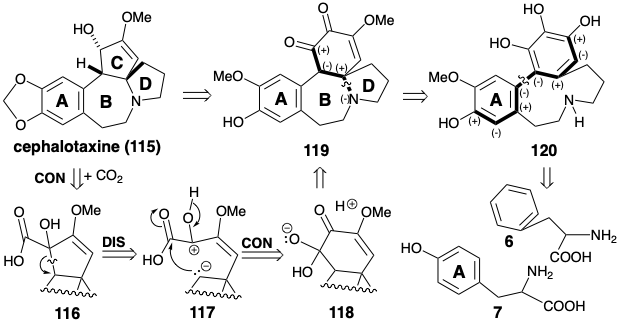
Ретросинтетично дислокація продукту перебудови бензилової кислоти 116 до попередника 119 відповідає полярному роз'єднанню мігруючого вуглецю як нуклеофілу, що призводить до окислення кінцевої міграції. Подальше з'єднання нуклеофільного мігруючого вуглецю в 117 призводить до зменшення міграційного походження в попереднику 118. Полярний аналіз 119 передбачає полярне відключення азоту як нуклеофіла від електрофільного вуглецю β до карбонільної групи. Полярний аналіз попередника 120 свідчить про те, що ароматичні кільця двох попередників 6 і 7 можуть бути з'єднані окислювальною муфтою.
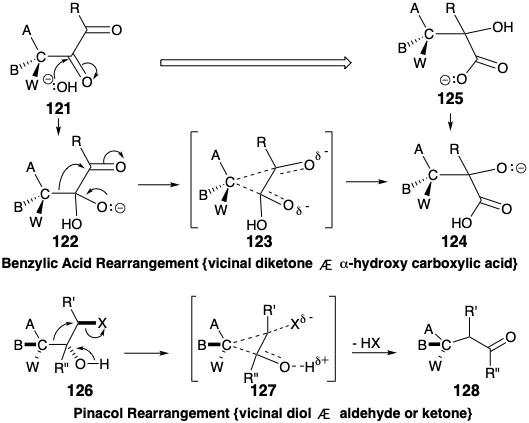
Послідовність з'єднання-відключення перестановки бензилової кислоти, узагальнена при перетворенні 121 на 125, механічно аналогічна перестановці пінаколів, розглянутої в розділі 4 (див. Розділ 3.4). Перестановка 122 на 124, що беруть участь в перестановці бензилової кислоти, є ізоелектронним з перетворенням 126 на 128 перестановки пінаколів, тобто ті ж електронні рухи в однакових масивах атомів, зв'язків і задіяні незв'язні електрони. Нуклеофільне додавання гідроксиду до α-дикетону 121 ініціює перестановку бензилової кислоти, яка протікає через тимчасово-мостовий перехідний стан 123, і в кінцевому підсумку виробляє α-гідроксикислоту 125. Перебудова пінаколів протікає через тимчасово-мостовий перехідний стан 127. Як у перебудовах бензилової кислоти, так і в пінакольних перебудовах мігруюча група діє як внутрішній нуклеофуж-нуклеофіл, який додає до електрофільного терміналу міграції. В обох перестановках рівень функціональності джерела міграції збільшується, а рівень функціональності кінцевої міграції зменшується.
Вважається, що біосинтез цефалотаксину (115) передбачає окислювальну зв'язок двох багатих електронами ароматичних кілець у фенетилізохіноліні 6 проміжного 128, що доставляє тетрациклічний Δ-аміно-α, β- ненасичений кетон 129. Полярне утворення та подальше полярне розщеплення тимчасового шестичленного гетероциклу азоту в 128 полегшує окислювальну зв'язок, роблячи його ентропічно більш сприятливим внутрішньомолекулярним циклогексаннеляцією, а не циклодеканнеляцією, яка повинна генерувати 130 безпосередньо. Доцільно постулювати наявність метоксильної групи в позиції 7 у 128, оскільки це може бути пов'язано з регіоселективним окислювальним зв'язком у положенні 8а, що є пунктом до гідроксильної групи, яка, як передбачається, присутня на позиції 6. Ця регіоселективність контрастує з тією, що спостерігається в окислювальному сполученні осінньої лінії (28) в положенні 4а (див. Розділ 6.1). Таким чином, О-метильні групи в 28 і 128 служать регіоконтрольними елементами в окислювальних муфтах цих фенетилізохіно-ліній. Редукція та регіоселективне метилювання 130 встановлюють стадію для регіоселективної електрофільної активації шляхом окислення ортогідрохінону 131 до орто-хінону 132.
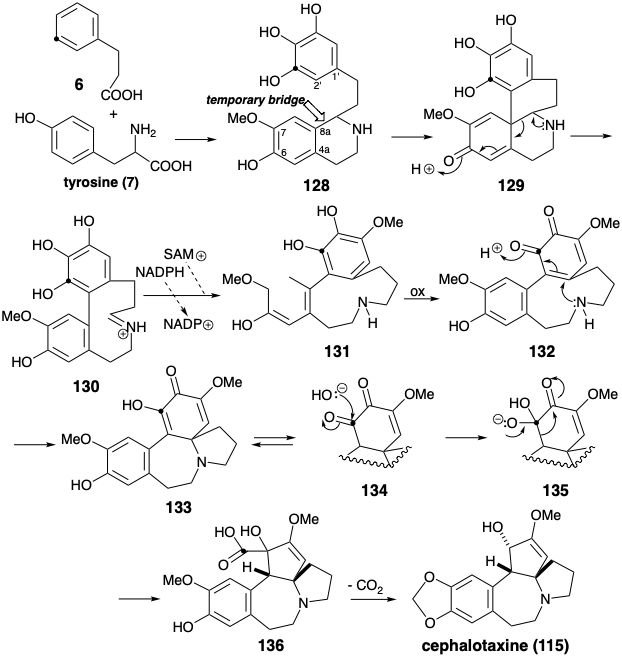
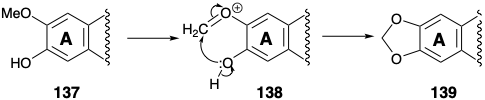
Нуклеофільний Michael додавання вторинного аміну потім доставляє 133 кето-таутомер яких 134 є α-дикетоном. Бензилова кислота перебудова, ініційована перетворенням на 135, забезпечує α-гідроксикислоту 136, в якій карбоксильний вуглець отримується з метавуглецю вихідного матеріалу фенілаланіну (6). Втрата цього вуглецю як вуглекислого газу потім генерує цефалотаксин (115) після перетворення масиву ортометокси-фенолу в групу метилендіоксидів. Це перетворення, тобто 137 до 139, є загальним у природі і, імовірно, включає окислювальну генерацію електрофілу 138.
B Кільцеве відпалювання електрофільним ароматичним заміщенням
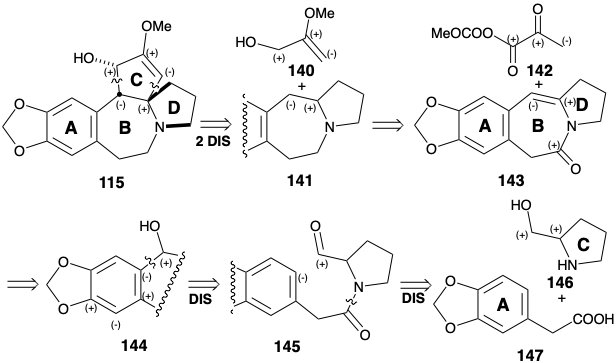
Як і в біосинтезі цефалотаксину (115), стабільність ароматичних похідних рекомендує використовувати ароматичний попередник для кільця А. стратегія Вейнреба для побудови скелета цефалотаксину7 визнає потенційну корисність аміногрупи та дисонантного С-кільця функціональність для активації полярних реакцій, які могли б додати C-кільце до попередника ABD-кільця. Синтетичні еквіваленти 142 і 143 відповідають полярним синтонам 140 і 141. Карбонільна група в 143 додається для полегшення генерації попередника 145, аміду пролінолу (146) та арилуксусной кислоти 147. Енамін у 143 може бути отриманий шляхом зневоднення попередника β-гідроксиаміну 144, який, в свою чергу, повинен бути доступний безпосередньо шляхом полярного об'єднання ароматичного нуклеофілу та альдегіду електрофілу в 145.
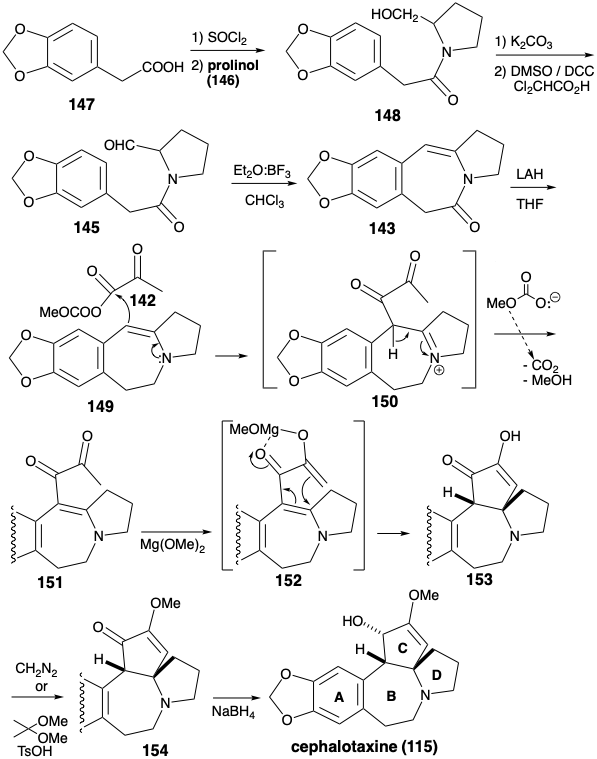
Енамін 143 був побудований шляхом відпалу кільця B між ароматичним кільцем A попередника 147 і попередньо сформованим кільцем D попередника 146. Маскування гідроксильної групи в 146 є непотрібним, оскільки ацилювання відбувається при більш нуклеофільному азоті, щоб дати амід 148, а не при менш нуклеофільному кисні для отримання ефіру. Остаточний зв'язок кільця В був утворений електрофільним ароматичним заміщенням, яке відбувалося виключно в менш перевантаженому аріловому положенні в 145. Відслуживши свою мету, амід карбоніл був відновно видалений з 143, щоб доставити 149. Полярна активація, яку надає група ацилів у 142 році, вперше використовується для об'єднання 142 та 149, щоб дати 150. Потім полярна активація, що надається обома карбонільними групами, експлуатується для завершення відпалу кільця С. Внутрішньомолекулярне додавання аніону енолату до електрофільного β-вуглецевого атома α, β-ненасиченої карбонільної системи призводить до 153. Необхідний цис-кільцевий сплав кільця С, безсумнівно, є найбільш стабільним. Особливо примітна група виходу метилкарбонатного аніона в 142 році. Декарбоксилювання цього аніону генерує метоксид in situ, який потім депротонує проміжний іон iminium 150 для отримання Michael акцептора 151 в виключно м'яких умовах. Також заслуговує на увагу використання метоксиду магнію в якості основи для генерації енолату 152 в синтезі Вайнреба цефалотаксину. Магній може допомогти циклізації шляхом хелатування, що забезпечує сприятливий цисоїдної конформації. Остаточне регулювання функціональності передбачало етерифікацію енолу і зниження гідридів. Доставка гідриду відбувається з менш стерильно перевантаженої опуклої грані 154, що утворює 115 з правильною відносною конфігурацією в третьому асиметричному центрі в кільці С. Отримано регіоізомерний еноловий ефір разом з 154. Цей ізомер може бути перероблений шляхом каталізованого кислотою рівноваги.
B Кільцеве відпалювання за допомогою нуклеофільної ароматичної заміщення
Тоді як відпалювання кільця В в синтезі Вайнреба цефалотаксину (115) було досягнуто електрофільним ароматичним алкілуванням, синтез Семмельхака створює такий же зв'язок нуклеофільним ароматичним алкілуванням. 8 Стратегія Semmelhack використовує карбоніл C-кільця для забезпечення необхідної нуклеофільності в кінцевому проміжному 155. Як і в біосинтезі та стратегії Вайнреба, ароматичний попередник використовується для кільцевого А. N-алкілювання CD-кільцевого амінного фрагмента 157 з фрагментом А-кільця 156 забезпечує 155. Один і той же вихідний матеріал А-кільця 147 використовується для обох тотальних синтезів. Кільце С в 157 містить дві функції кисню, які забезпечують електрофільну активацію на відповідних атомах вуглецю. Таким чином, ці функціональні групи не можуть бути використані безпосередньо для створення зв'язку між цими атомами вуглецю полярною реакцією. У синтезі Вайнреба цефалотаксину (115) кільце С було побудовано полярними реакціями за допомогою вихідного матеріалу 142, який включає дисонансний контур між двома функціями кисню в С-кільці. Стратегія Semmelhack визнає, що ця дисонансна схема в 157 може бути утворена неполярною реакцією, відновним зв'язком двох електрофільних карбонільних вуглеців у попереднику 158. Хоча 158 може бути доступний безпосередньо шляхом полярного додавання двох карбометоксиметилових нуклеофілів до електрофільного попередника D-кільця 160, Semmelhack вибрав альтернативну стратегію додавання двох алільних нуклеофілів до 160 з подальшим окислювальним розкриттям латентні карбоксильні групи в проміжному 159.
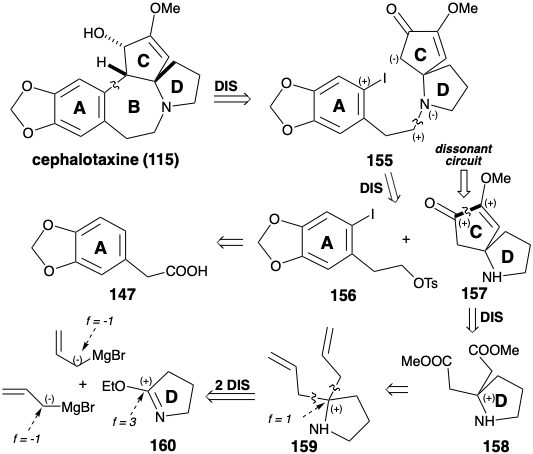
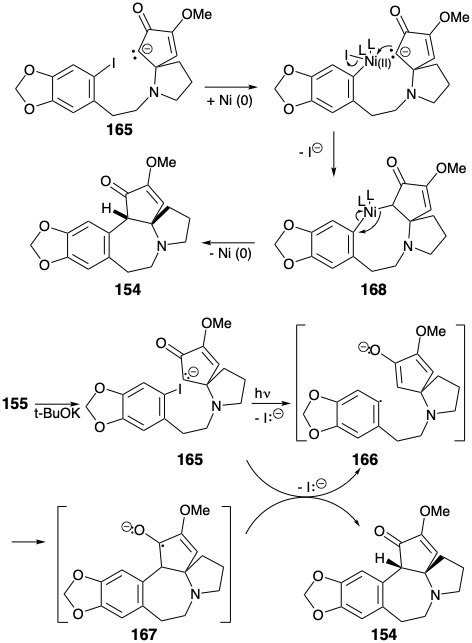
CD-кільце проміжного 157 готували з піролідинону. Реакція ефіру іміно 160 з алліловим нуклеофілом дала 159. Маскування аміногрупи як кислотного лабільного аміду 161 було потрібно до окислювального розщеплення C-C π-зв'язків у 159, що в кінцевому підсумку забезпечило діефір 158. Внутрішньомолекулярне ацилоїнове з'єднання 158 у присутності хлортриметилсилану (модифікація Ruhlmann) вироблялося 162, який окислювався безпосередньо до 163 шляхом додавання брому в один горщик і елімінації TMSbr. Метилювання цього симетричного діону доставлено 157. Алкілювання цього аміну нітрозилатом 164 передбачено 155. Циклізація 155, vide infra, з подальшим зменшенням проміжного 154 доставленого цефалотаксину (115). Ця синтетична стратегія призводить безпосередньо до правильного енолового ефіру 154 без утворення регіоізомерного енолового ефіру, який є побічним продуктом синтезу Вайнреба.
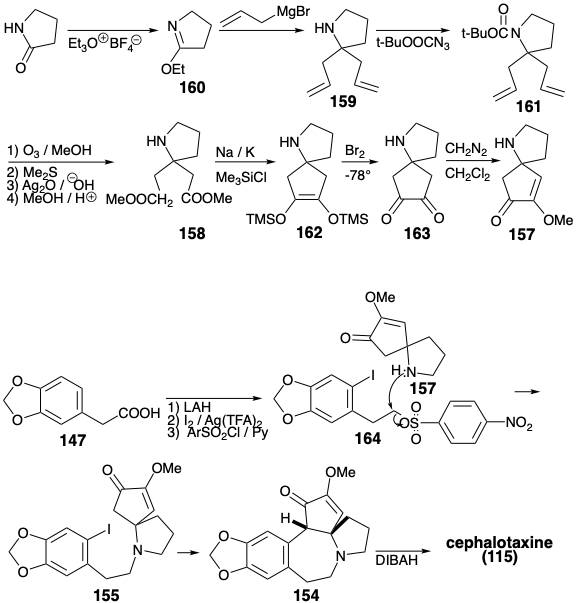
Ключова циклізація від 155 до 154 була досягнута різноманітними реакціями, що включають нуклеофільну ароматичну заміщення. Звичайно, пряма нуклеофільна атака на багате електронами кільце аріл йодид не відбувається, коли нуклеофіл енолатів генерується з 155. Однак чисте нуклеофільне заміщення може бути здійснено фотолізом енолату 165 або обробкою 165 Na/K або нікелевим (0) каталізатором. Найкращі виходи продукту циклізації 154 (94%) були отримані фотостимульованою реакцією S RN 1 імовірно за участю ланцюга, що несе аніонні радикали 166 і 167. Реакція S RN 1 також може бути досягнута (45%) шляхом реакції енолату 165 з Na/K. нікель (0) -каталізована реакція 165 за умови 154 в 30% вихід імовірно шляхом окислювального додавання арілгалогеніду до Ni (O) та нуклеофільної заміщення йодиду карбоніоном, що виробляє σ -арил-нікель (II) проміжний 168, який піддається відновному ліквідації 154 для регенерації каталізатора Ni (0).