6.7: Лікоподин
- Page ID
- 18618

Біосинтез алкалоїдів з L-лізину
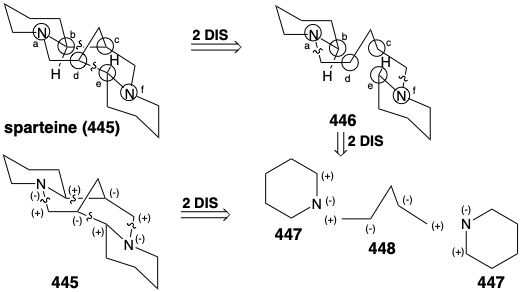
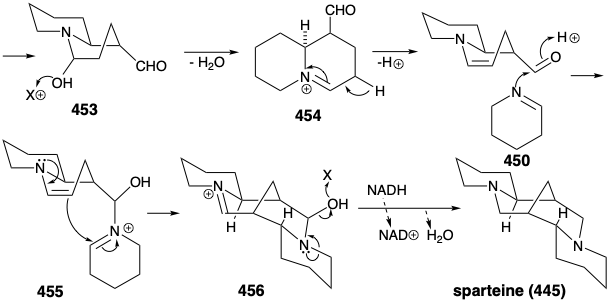
Різноманітні топологічно складні гетероцикли насиченого азоту побудовані в природі з простих ациклічних попередників. Різка логіка цих біосинтезів особливо вражає, якщо розглядати або з точки зору топологічної, або полярної реактивності. Наприклад, примітна ефективність, з якою зібраний хитромудрий багатоциклічний скелет спартеїну (445), виключно з трьох молекул симетричного синтетону. Топологічний аналіз 445 виявляє наявність шести загальних атомів a-f. Розщеплення двох зв'язків між двома парами загальних атомів, b-c і d-e, спрощує топологію до двох піперидинових кілець, з'єднаних прямим ланцюгом в 446 році. Цей проміжний продукт легко отриманий з двох п'яти вуглецевих синтезонів 447 і 448. Аналіз полярної реактивності 445 показує, що полярні реакції, активовані аміногрупами в 445, повинні легко дозволити його будівництво з 447 і 448.
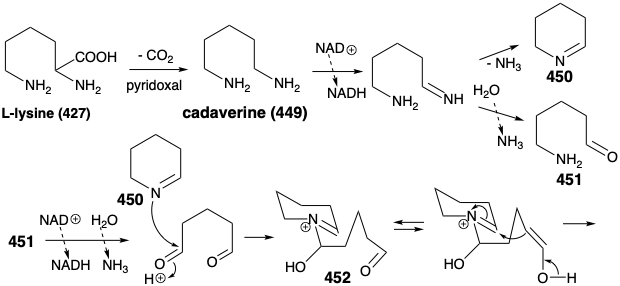
Насправді функціоналізовані синтетичні еквіваленти як для 447, так і 448 готуються в природі з L-лізину (427). Таким чином, піридоксальне каталізоване декарбоксилювання 427 виробляє симетричний діамін 449. Окислення і гідроліз 449 через 451 дозволяють собі пентандіальний, який забезпечує похідне імінію 452 шляхом реакції з 450. Внутрішньомолекулярна конденсація альдолу тоді забезпечує 453. Похідне імінію 454 від дегідратації 453 дає похідне імінію 455 шляхом реакції відповідного енаміну з другим еквівалентом imine 450. Друга внутрішньомолекулярна конденсація альдолу забезпечує 456. Зневоднення і зниження забезпечує спартеїн (445).
Біосинтез лікоподину
Ми бачили, що багато натуральних продуктів утворюються з одного вихідного матеріалу, таких як (а) багато полікетідів, жирних кислот або простагландинів з ацетил КоА, (б) багато алкалоїдів з шикімової кислоти, або (в) терпени з мевалонової кислоти. Однак деякі натуральні продукти утворюються змішаними біосинтезами з комбінацій цих вихідних матеріалів. Таким чином, лізергінова кислота (див. Розділ 6.4) виникає з хоризмової кислоти плюс мевалонової кислоти ізопентенілпірофосфат плюс цукор, D-рибоза. Аналогічно алкалоїди індолу (див. Розділ 6.5) виникають з хоризмової кислоти плюс терпену, похідного мевалонової кислоти, секологанін, плюс цукор, D-рибоза.
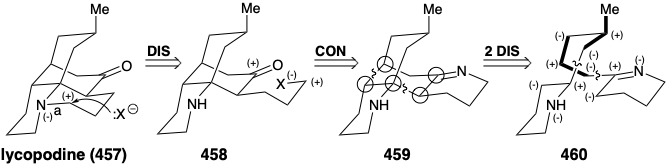
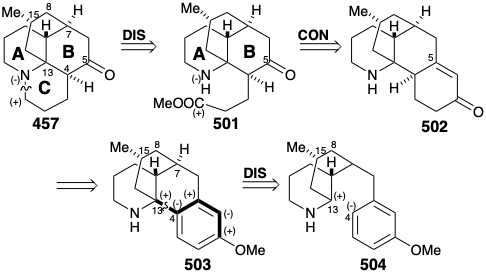
Тепер ми побачимо, що мостовий багатоциклічний скелет алкалоїду лікоподину (457) виникає з ацетоацетилу CoA плюс L-лізину (427). Що стосується спартеїну вище, то як топологічний, так і полярний аналіз біосинтетичної стратегії для 457 виявляють його різку логіку. Карбонільна група в 457 році генерується в природі сольволітичним розщепленням тимчасового моста. У процесі генерується пропілзамінник з електрофільною активацією в кінці, який потім використовується для побудови кінцевого кільця 457. Ретросинтетично це передбачає відключення до 458 з подальшим повторним підключенням до 459. Значне спрощення цієї субмети є результатом роз'єднання двох зв'язків між парами загальних атомів у 459 до 460. Полярний аналіз 460 показує, що відновлення цих зв'язків може бути досягнуто шляхом використання полярної активації, що забезпечується атомами азоту в 460. Крім того, 460 можуть бути зібрані з двох великих фрагментів полярною реакцією, що утворює будь-який із зв'язків у вуглецевому ланцюзі, що з'єднує два гетероцикли азоту.
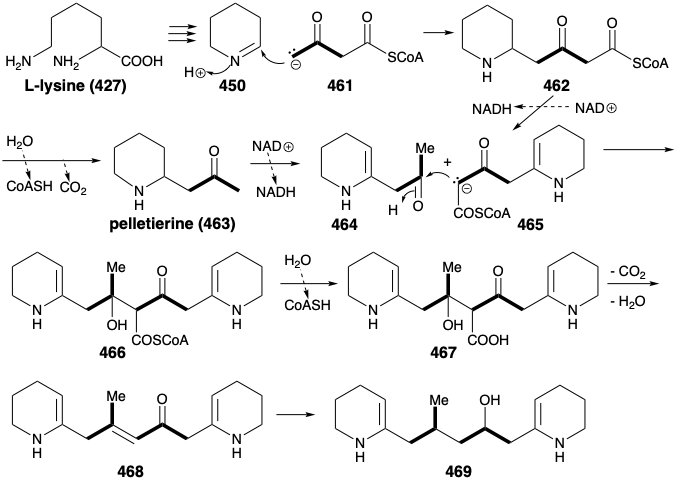
У природі два кільця піперидину в 460 походять від L-лізину (427), а з'єднувальний ланцюг зібраний з двох трьох вуглецевих одиниць, отриманих з актетоацетилу CoA (461). Конденсація Aldol 461 з 450 дозволяє 462. Зверніть увагу, що алкілування 461 відбувається при менш кислому δ вуглецю. Можливо, це стосується пов'язаного з ферментом похідне енаміну 470 (див. Нижче) з 461. Окислення та депротонування 462 забезпечує 465, тоді як 462 також дає гранулят (463) шляхом гідролізу та декарбоксилювання. Конденсація Aldol між 464 і 465 потім забезпечує 466, тобто гідролізується до 467. Декарбоксилятивна елімінація дає 468, тобто зменшується, щоб забезпечити 469, синтетичний еквівалент синтону 460, сформованого в стратегічному аналізі, представленому нижче.
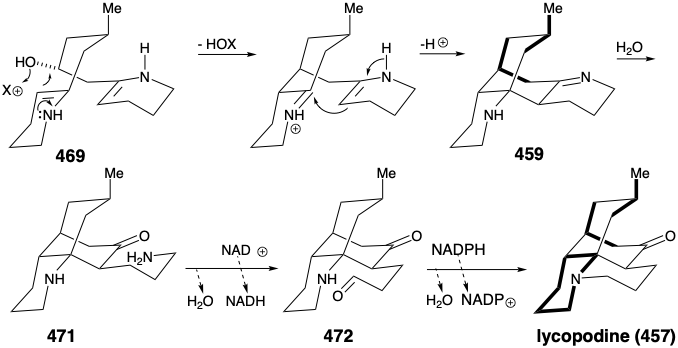
Циклізація 469 шляхом внутрішньомолекулярного алкілування енаміну з подальшою внутрішньомолекулярною альдоподібною конденсацією виробляє проміжний 459, запропонований у стратегічному аналізі. Гідроліз іміну в 459 утворює карбонільну групу, необхідну для лікоподіну. Окислення отриманого пропіламіну 471 до альдегіду 472 з подальшим внутрішньомолекулярним відновним алкілуванням потім виробляє лікоподин (457), в якому одне кільце піперидину, отримане лізину, чітко помітне, тоді як два полікетидні отримані ацетонільні одиниці та п'ять вуглецеві одиниці з другої молекули лізину складні переплітаються між собою.
Смертельно недосконала стратегія синтезу лікоподину
Ми бачили, що як топологічний, так і полярний аналіз біосинтетичної стратегії лікоподину (457) висвітлюють логіку процесу. Дві функціональні групи в 457 році можуть бути використані в різних стратегіях для полегшення побудови скелетної мережі за допомогою полярних реакцій. Є п'ять загальних атомів в 457, чотири атоми вуглецю, які знаходяться в кільці B, і азот. Спочатку ми розглянемо смертельно недосконалу стратегію, яка генерує лише епімер, а не сам натуральний продукт.
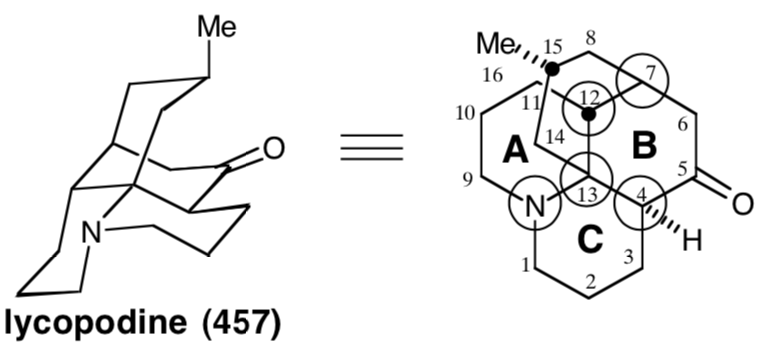
Полярний аналіз кільця B показує, що полярні реакції, що використовують полярну активацію, що забезпечується аміно-та карбонільними функціями в 457 році, можуть бути використані для побудови будь-якого зв'язку цього кільця. У підході Віснера до лікоподин,19 утворюється остаточний скелетний зв'язок між загальним атомом 7 і непоширеним атомом 6, що відповідає дислокації від 457 до 473. Утворилася передостання зв'язок полягає в тому, що між загальними атомами 4 і 13, що відповідає дислокації 473 до біциклічного попередника 474. Елегантність стратегії полягає в плані здійснення циклічної проміжної 474 до тетрациклічного скелета цілі 457 за один крок. Синтетичний еквівалент 475 з 474 має додаткові карбонільні групи на позиціях 7 і 9. Перший забезпечує додаткову електрофільну активацію при С-7, а другий деактивує нуклеофільність аміногрупи. Біциклічний проміжний 475 може бути доступний з симетричного моноциклічного попередника 476. Початкова аміногрупа 457, нітрильний азот у 476, навіть забезпечує полярну активацію для побудови 476 з акрилонітрилу та дигідрорезоціналу.
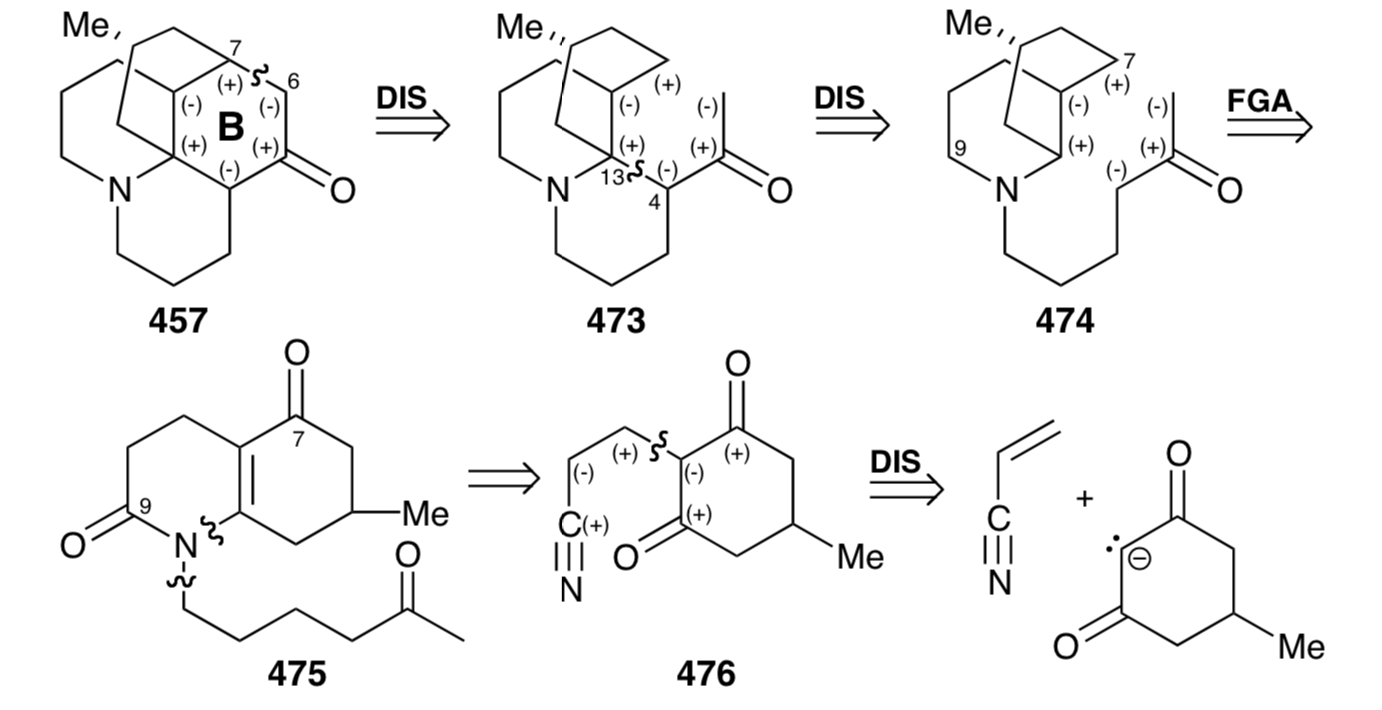
Фактично, каталізований кислотою гідроліз 476 призводить безпосередньо до енаміду 477, який забезпечив 475 N- алкілування алкілбромідом 467 і подальше гідролітичне видалення маскуючої металевої групи. Базово-каталізована внутрішньомолекулярна реакція Майкла 475 може генерувати два стереоізомери в положенні 13, які є результатом додавання карбоніону до будь-якої грані D-кільця в 479. Однак, як і очікувалося, стеричний контроль підходу сприяє стереоселективному додаванню на стороні D-кільця навпроти метилового замінника, щоб дозволити собі проміжний 481, а не небажаний стереоізомер 480. Тим не менш, синтез смертельно недосконалий, оскільки подальша реакція альдолу дала виключно 484, скелет якого епімерний з лікоподином при С12. Таким чином, С-12 в проміжному 481 є епімеризуемий, а епімер 482 мабуть циклізується в повній перевазі 481. Це виробляє 484, а не 483, що потрібно для синтезу лікоподину (457). Редуктивне видалення амідних карбонільних і третинних гідроксильних груп з 484 доставлено 12-епі-лікоподин (485).

Релейна стратегія та симетричний попередник для лікоподину
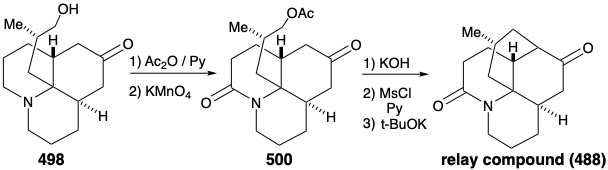
Друга стратегія синтезу лікоподину генерує кільце D шляхом циклізації трициклічного синтеону 486 з попередньо сформованими кільцями AB і C. 20 Ця стратегія була спрямована перспективою використання симетричного плавленого трициклічного кетону 487 як вихідного матеріалу. Таким чином, топологічний аналіз 457 рекомендує відключення двох зв'язків між загальним (обведеним) і непоширеним атомом, зв'язками 7-8 і 13-14, щоб повністю видалити D-кільце. Супутня транспозиція карбонільної групи з С-5 в 457 до С-6 потрібна для генерації симетричного попередника 487. Крім того, ця транспозиція в 486 дозволяє формувати зв'язок 7-8 C-C скелета лікоподіну шляхом внутрішньомолекулярного алкілування, яке використовує полярну активацію, що забезпечується карбонілом у положенні 6. Тоді як азот у 486 може забезпечити електрофільну активацію для формування зв'язків С-С у положенні 13 в 487. Амід карбоніл при С-9 в 486 включений як деактивуюча група для зменшення нуклеофілічності аміногрупи, що знешкоджує небажану кватернізацію, яка може конкурувати з алкілуванням нуклеофіла карбоніону при С7.
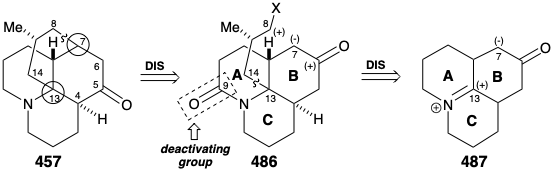
Внутрішньомолекулярне алкілювання кетону 4 86 дасть 488. Було б заспокійливим, якби заключні кроки синтезу можна було б розробити за допомогою зразка 488, який можна було б легко приготувати з натурального продукту 457, можливо, через діол 489, який вже був підготовлений з 457 під час структурних досліджень на алкалоїди лікоподію. Цей зразок з'єднання 488 потім можна було б використовувати замість синтетичного матеріалу для опрацювання деталей перетворення 488 на 457. Це ще один приклад стратегії, відомої як релейний підхід, який ми бачили, що використовується в синтезах еритроноліду B (див. Розділ 5.4) та хініну (див. Розділ 6.5). Перевага такого підходу полягає в тому, що цінний ключовий проміжний продукт можна отримати легко в кількості. Релейна сполука (наприклад, 488) стає мішенню синтезу.
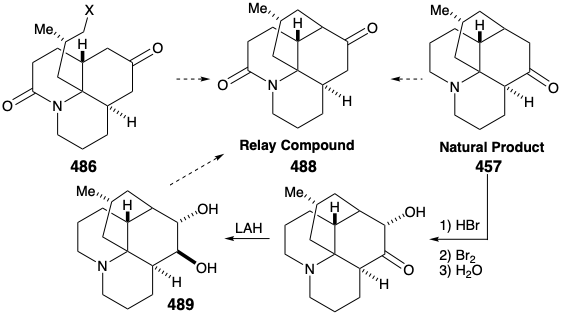
Розглянемо спочатку взаємоперетворення релейного з'єднання 488 і 457, перш ніж досліджувати сумарний синтез релейного з'єднання. Для диференціації гідроксилів в положеннях 5 і 6 діол 489 від природного лікоподину (457) був моноацетильований на стерильно найбільш доступному гідроксилі С-6. Зневоднення з подальшим гідролізом дозволило 490. Окислення аллільного гідроксилу з подальшим відновленням одержуваного α, β-ненасиченого кетону і перманганату окислення α до третинного аміну дало запропоновану релейну сполуку, амід 488, в 13% загального виходу з природного лікоподину (457).
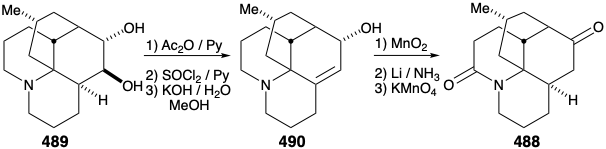
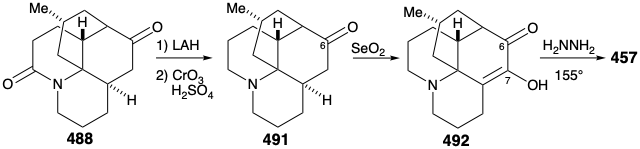
Потім реконверсія релейної сполуки 488 в лікоподин (457) була досягнута шляхом видалення карбонілу аміду шляхом відновлення за допомогою LAH та окислення одержуваних епімерних спиртів С-6. Потім амінокетон 491 окислювався до диосфенолу 492, який був зменшений вибірково реакцією Вольфа-Кішнера з гідразином гідратом, щоб дозволити собі лікоподин (457).
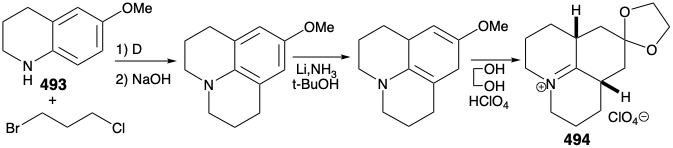
Як зазначалося вище, передбачався синтез 486, а значить і релейного з'єднання 488, з симетричного вихідного матеріалу (див. Вище). Зокрема, 486 може бути отриманий з 487 шляхом реакції з нуклеофільним синтоном бічного ланцюга. З положенням маскування електрофільності карбонільної групи ця стратегія виявилася життєздатною. Так, 494 отримано з таліну (493) шляхом алкілування 1-бром-3-хлорпропаном з подальшим розчиненням відновлення металу і кеталізацією. Реакція 494 з нуклеофільним фрагментом 495 з подальшим гідролізом дає цис-сплавлений трициклічний амін 496. Закриття кільця цього епімера неможливо. Епімеризація до транс, цис ізомеру 485 повинна передувати замиканню кільця. Таким чином, епімеризація була здійснена шляхом використання карбонільної функціональності в 496 шляхом бромування з подальшим дегідробромізації Маттокса-Кендалла і розчиненням відновлення металу. Деметилювання продукту 497 тоді дозволило суміш рацемічних діастереомерних кетонів 498 і 499. Вони були розділені хроматографією на глиноземі. Мінорний ізомер 498 володів природною відносною конфігурацією метилзамінника. Таким чином, даний синтез є нестереоспецифічним, і майже фатальна велика втрата цінного матеріалу відбувається внаслідок утворення небажаного стереоізомеру 499.
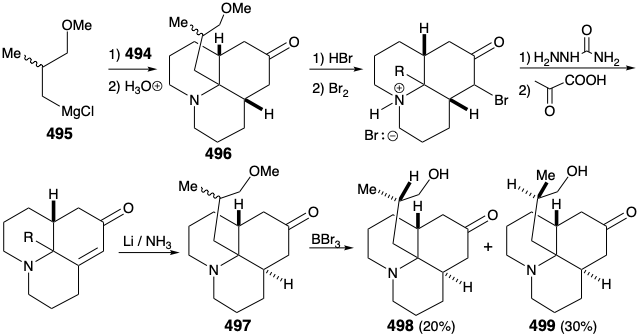
Перш ніж внутрішньомолекулярне алкілювання трициклічного кетону може бути здійснено, нуклеофільність аміногрупи повинна була бути ослаблена шляхом перетворення аміногрупи в 498 в амід 500, щоб уникнути N-алкілування. Омилення, мезилювання та внутрішньомолекулярне алкілювання потім забезпечили релейну сполуку 488 у рацемічній формі. Оскільки 476, хоча і гомохіральний, отриманий з природного лікоподину (457), вже був перетворений на 457, як обговорювалося вище, загальний синтез був завершений.
Ненавмисно біоміметична стратегія
У двох стратегіях для лікоподину (457), розглянутих вище, одне згенероване кільце B останнє та одне генерується кільце D останнім. Зараз ми розглянемо стратегію, яка генерує кільце С останнім, як виявлено в біосинтезі 457. Більше того, в подальшій аналогії з біосинтетичною стратегією, три вуглецевого ланцюга замінника на кільці B 501, тобто використовується для завершення кільця С, включений у тимчасове кільце в попереднику 502 шляхом приєднання до (прихованого) карбонільного вуглецю при С-5. Нарешті, приголосний контур між аміно- та метоксичними групами в ароматичному попереднику 503 передбачає полярну конструкцію з 504 зв'язку 4-13 C-C, знову ж таки за аналогією з біосинтетичною стратегією, шляхом атаки електрофілу С-13 на нуклеофільний центр на початковому С-4 . Однак ця стратегія була задумана до того, як було з'ясовано біосинтез лікоподину. За словами автора стратегії, «хоча конкретний синтетичний план, який дотримувався для побудови тетрациклічної системи, не мав особливої основи в біогенетичних міркуваннях, зовсім недавня робота запропонувала біогенетичний шлях, в якому найважливіший крок циклізації разюче схожий на той, який ми розробили». 21
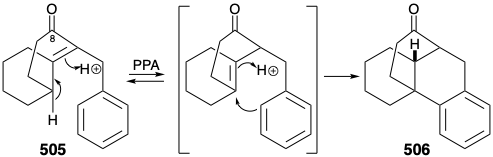
Перш ніж розглядати успішну реалізацію цього плану, повчально відзначити, що він був розроблений за допомогою уроків, засвоєних під час спроб досягти синтезу з використанням інших, смертельно недосконалих, стратегій. Один невдалий ранній план трансформації, аналогічний поколінню 503 з 504, був підтриманий успішними модельними дослідженнями. Так, карбоциклічна модель 505 охоче піддавалася аналогічній циклізації 506 при нагріванні поліфосфорною кислотою. Передбачалося, що метиловий замінник у положенні 15 на D-кільці 504 може бути введений згодом шляхом використання нуклеофільної активації, що забезпечується карбонільною групою на позиції 8. Крім того, включення цієї карбонільної групи може підвищити загальну корисність синтезу, оскільки заміщення кисню знаходиться на позиції 8 у багатьох алкалоїдах лікоподію. Однак, на відміну від карбоциклічної моделі 505, циклічності гетероциклічного аналога 507 дати 508 не вдалося. Швидше 507 був, мабуть, схильний до усунення, що призводить до ароматизації.
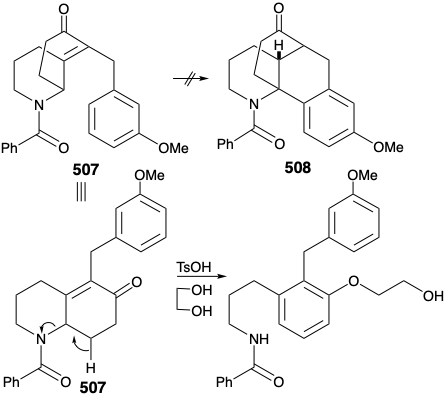
Тим не менш, обнадійливе спостереження з'явилося в результаті цього модельного дослідження. Таким чином, диалкілірованное похідне 509, побічний продукт в синтезі 507, зазнало бажаного типу циклізації. Успіх цієї циклізації, здавалося, пояснюється двома факторами, осьовою орієнтацією бензилового замінника та блокуванням шляху елімінації.
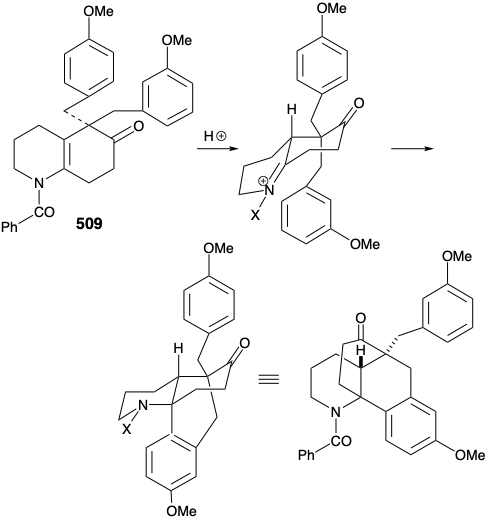
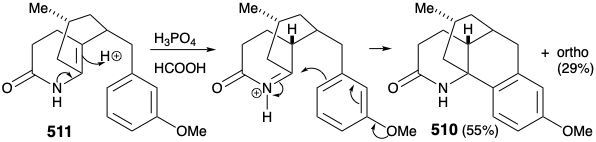
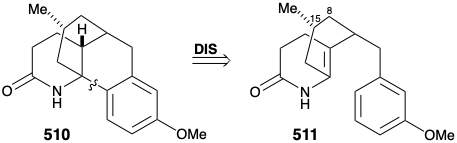
Потім була розроблена модифікована стратегія, в якій метиловий замінник, необхідний у положенні 15, був введений до відпалу кільця B, а карбонільна група в положенні 8 була видалена. Більше того, стереохімія цього метилзамінника в лікоподині диктувала транс-зв'язок метильної та м-метоксибензилової груп у новій субмішені. Функціоналізована похідна, обрана для втілення цих вимог, становила 511. Далі було визнано, що транс-метиловий замінник у 511 повинен практично усунути енергетичний бар'єр для досягнення осьової орієнтації бензилового замінника, необхідного для циклізації до 510.
Було досліджено декілька стратегій синтезу субмети 511, що, очевидно, є похідним з 512. Підхід до 512 шляхом кон'югатного додавання метилового нуклеофіла до 513 з подальшим окислювальним розщепленням циклогексенону 514 був виключений схильністю 514 до ізомеризації в β, γ-ненасичений ізомер 515.

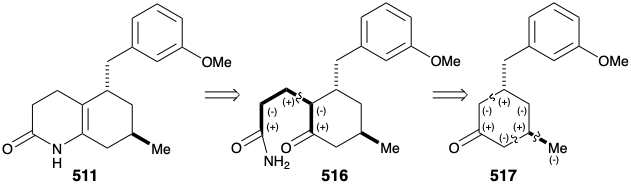
Другий підхід до 511 використовує приголосний контур між карбонільним вуглецем та енамідним азотом у 511 або пов'язану з нею контур приголосних між двома карбонільними групами в кето-аміді 516. Відключення трьох вуглецевих бічних ланцюгів передбачає попередник циклогексанону 517, який може генеруватися полярними з'єднаннями, активованими карбонільною групою.
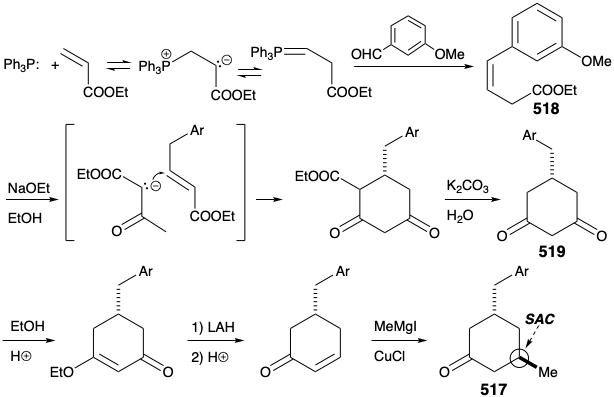
Циклогексанон 517 був отриманий шляхом полярних реакцій між акриловим ефіром і ацетооцтовим ефіром. Таким чином, симетричний діон 519 був утворений з β, γ-ненасиченого ефіру 518 шляхом прототропної аллілільної перестановки з подальшим додаванням Майкла етилацетоацетату, циклізації Дікмана, гідролізу та декарбоксилювання. Селективному відновленню тільки однієї карбонільної групи в 519 році сприяло маскування другого карбонілу цього діону як вінілового ефіру. Потім кислотно-каталізоване зневоднення забезпечило циклогексенон, який доставляв 517 при стереоселективному 1,4-додаванні метилнуклеофіла.
Оригінальна стратегія побудови ключового проміжного 511 з циклогексанону 517 мала один серйозний недолік. Таким чином, перетворення 517 в 511 вимагає регіоселективного алкілування Михайла. Однак алкілування 517 відбувалося нерегіоселективно на обох вуглецях α до карбонілу, що утворює суміш 511 і 520.
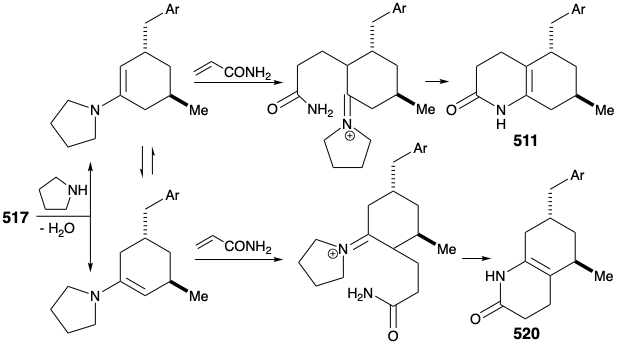
Остаточно розроблений вдосконалений, повністю структурно специфічний синтез субмішені 511, який експлуатував регіоспецифічну генерацію та електрофільну ловлю енолату 522. Це було досягнуто шляхом додавання Михайлом бензилового нуклеофіла до циклогексенону 521. Отриманий регіоспецифічний енолат потім алкілували аллілбромідом. Процес також є високостереоселективним завдяки перевазі осьової атаки в Michael доповнень органомідних нуклеофілів та перевагу екваторіальній диспозиції метилзамінника в 521 році. Крім того, необхідний транс-зв'язок між аллілом та бензиловим замісниками забезпечується термодинамічним контролем завдяки епімеризуваності α до кетонового карбонілу. Гідроборація та окислення аллільного бічного ланцюга, етерифікація та реакція одержуваного кетоефіру з аміаком забезпечили ключовий проміжний продукт 511.
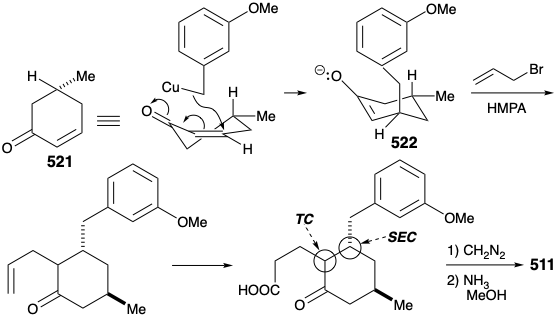
Внутрішньомолекулярне електрофільне ароматичне заміщення дало переважно бажаний паразаміщений продукт циклізації 510 (55%) разом з деяким продуктом ортозаміщення (29%). Потім амідний карбоніл був видалений відновленням з LAH, а захисна ароматичність арилового кільця була видалена відновленням берези. План впливу кільцевого розщеплення шляхом окислення α, β-ненасиченого циклогексенону не вдалося через термодинамічну перевагу необхідному енону 502 існувати як відповідний β, γ- ненасичений таутомер 524.
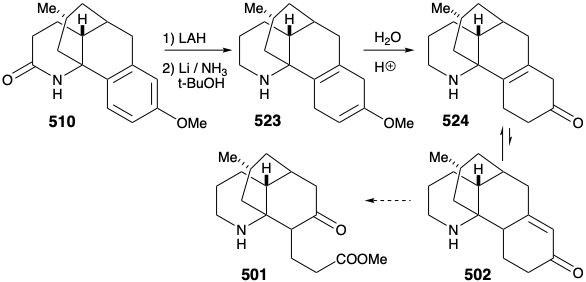
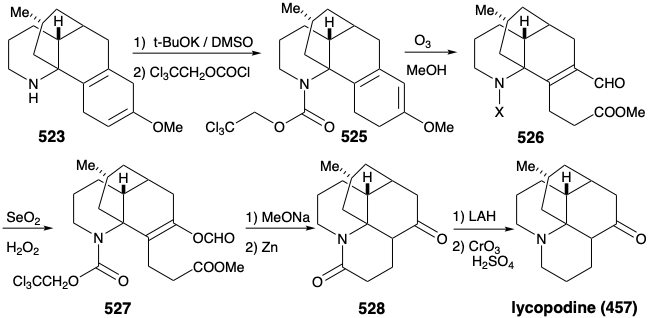
Тому ще раз довелося сформулювати альтернативну стратегію. Початковий план генерації 489 з 497 був смертельно недосконалим. Дійсно, хоча кожен крок у початковому плані був добре прецедентним, так була ймовірність того, що 502 буде в рівновазі зі значною сумою 524. Дійсно, ця ж проблема зірвала спробу синтезу 512 з 514 (див. Вище). Як це часто буває, недолік відомої методології досягнення важливої синтетичної мети, особливо якщо вона перешкоджає укладенню амбітного тотального синтезу, надихає застосування нової хімії для забезпечення рішення диллеми. Необхідність - мати винаходу! Таким чином, генерація 501 з 523 вимагала окисного розщеплення двох зв'язків, «а» і «б», в циклогексадіеновому кільці. Початковий план передбачав розщеплення зв'язку «а» спочатку після ізомеризації, яка розмістила легко розщеплюється зв'язок C = C в цьому положенні. В альтернативній стратегії зв'язок «b» спочатку розщеплюється після ізомеризації, яка розмістила легко розщеплюється зв'язок C = C в цьому положенні.
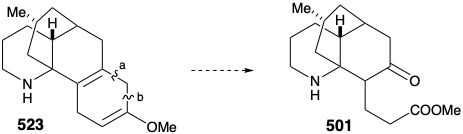
Таким чином, 1,4-циклогексадієн 523 був ізомеризований до кон'югованого 1,3-дієну, а аміногрупа була замаскована для захисту від окислення. Селективний озоноліз більш багатих електронами зв'язку C = C в 525 тоді дозволив альдегідометиловий ефір 526. Незвичайне окислення Bayer-Villager 526 дало енол формат 527, який дозволив кето-амід 528 після метанолізу ефіру енолу, видалення захисної групи карбамату з аміноазоту та лактамізації. Потім амід карбоніл був видалений, щоб забезпечити лікоподин (457) шляхом відновлення з LAH з подальшим повторним окисленням гідроксилу С-5 до необхідної карбонільної групи С-5.