5.4: Еритронолід B
- Page ID
- 18751

Фермент сприяє реакції пропіонілКоА з метилмалонілКоА генерує 83 енантіоселективно. Потім діастереоселективне відновлення забезпечує b-гідрокситіоефір 84. На відміну від біосинтезу жирних кислот, зневоднення і кон'югатного відновлення отриманого α, β-ненасичений тіоефір не передує конденсації Клайсена з другим еквівалентом метилмалонілКоА в процесі біосинтезу попередника секо кислоти 87 макроліду 6-дезоксиеритроноліду В.
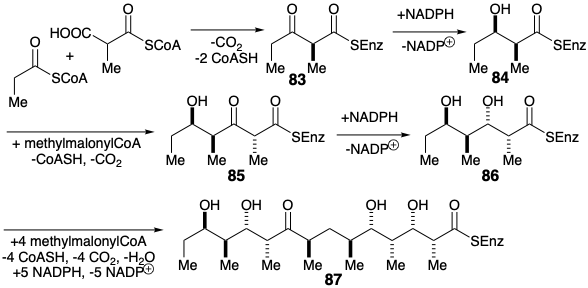
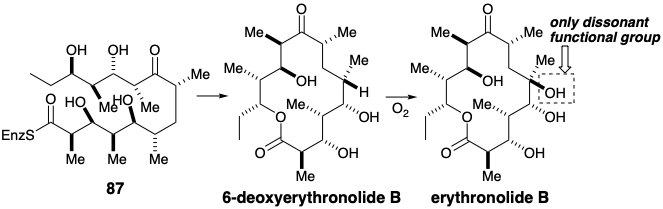
Отриманий β-кетотіоефір 85 зменшується до β-гідроксиефіру 86, який реагує з додатковими метилмалонілCOA та NADPH для отримання 87. Лактонізація потім забезпечує 6-дезоксиертронолід В, з якого еритронолід утворюється окисленням. Генерація лише 1 з 2048 можливих діастереомерів ациклічного проміжного продукту 87 є чудовим наслідком асиметрії гомохіральних каталізаторів (ферментів), які сприяють конденсаціям та скороченню, відповідальним за створення десяти асиметричних центрів.
Релейно-спрямована стратегія повного синтезу еритроноліду Б.
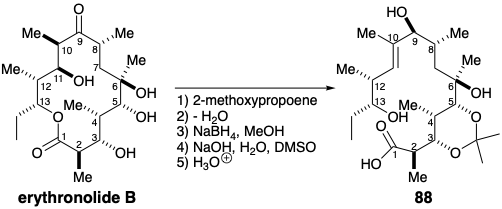
У техніко-економічному обґрунтуванні, природно отриманий еритронолід B був перетворений в ациклічну гідроксикислоту 88, яка використовувалася для розробки заключних етапів для загального синтезу. 12 Потім процес синтетичного проектування був спрямований вибором цього природним попередником, релейної сполуки. Два стереоцентри на позиціях 12 і 13 в 88 віддалені від тих, на позиціях 2-6 і 8. Тому було б важко генерувати один набір стереоцентрів під стереоконтролюючим впливом іншого. Швидше за все, будівельні блоки, що містять ці стереоцентри з правильними абсолютними конфігураціями, можуть бути зібрані, а потім об'єднані для забезпечення релейного з'єднання 88. Перший загальний синтез ерітроноліду B1 3 прийняв конвергентну абсолютну асиметричну (див. Розділ 4.6) стратегію, покликану забезпечити 88 шляхом об'єднання двох гомохіральних сегментів, нуклеофіла 89 та електрофіла 90.
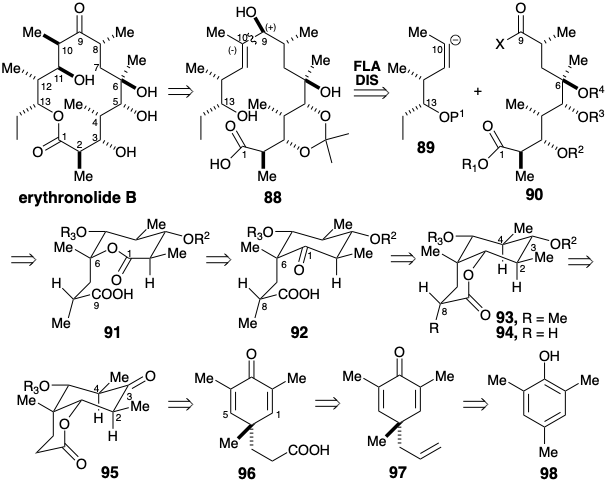
Для забезпечення більш конформально жорсткої платформи для генерації та підтвердження відносних конфігурацій стереоцентрів для ациклічної субмети 90 було передбачено циклічний тимчасово-мостовий попередник 91. Ще більшу жорсткість забезпечує менше кільце шестичленного кетону 92, який включає в себе функціональність лактону в прихованій формі. Можна очікувати, що стереоелектронно сприяє осьовій доставці водню до ізомерного циклогексанону 94, щоб генерувати екваторіальний гідроксил у положенні 3. Крім того, дислокація 93 до 94 дозволяє карбонілу в положенні 3 сприяти термодинамічно сприятливій екваторіальній диспозиції метилів у позиціях 2 і 4, і встановлює основу для виявлення симетричного попередника, Vide infra. Інший тимчасовий міст, шестичленний лактон може бути використаний для сприяння генерації необхідної конфігурації у відносно віддаленому стереоцентрі на позиції 8 в 92 під час метилювання 95, що, як очікується, сприятиме екваторіальному метилу в 94. Лактонні мости також можуть бути використані для забезпечення належної стерео- та регіохімічної орієнтації під час введення кисневих замінників у положеннях 1 та 6 шляхом полярних доповнень до зв'язків C = C в симетричному попереднику діенону 96. Пошук літератури доступних вихідних матеріалів з вуглецевим скелетом 96 може бути використаний для ідентифікації аллілциклогексадіенону 97, який легко готується з триметилфенолу 98.
Реакція з аллілбромідом фенолату від 98 забезпечує 100% перестановку Клейзеном вихідного продукту O-алілювання 99. Кислотно-каталізована перебудова Cope 100 забезпечує симетричний діенон 97, який вибірково гідроборується в кінцевій вініловій групі, а потім окислюється з отриманням карбонової кислоти 96. Стерео- і регіоселективна доставка кисню в положення 5 здійснюється шляхом внутрішньомолекулярного приєднання карбоксилату до зв'язку C = C в 96. Захоплення оборотно сформованого енолатного карбоніону проміжного продукту з електрофільним бромом виробляє 101. Щоб повторити цей процес на решті зв'язку C = C, лактон омилюється для регенерації карбонової кислоти. Це також генерує епоксид шляхом внутрішньомолекулярного зміщення бромозамісника алкоксидом. Друга стерео- і регіоселективна доставка кисню, на цей раз до позиції 1, знову здійснюється шляхом внутрішньомолекулярного додавання карбоксилату до зв'язку C = C. Подальше редуктивне видалення непотрібної функціональності гетероатома на позиціях 1 і 4 забезпечує 103.
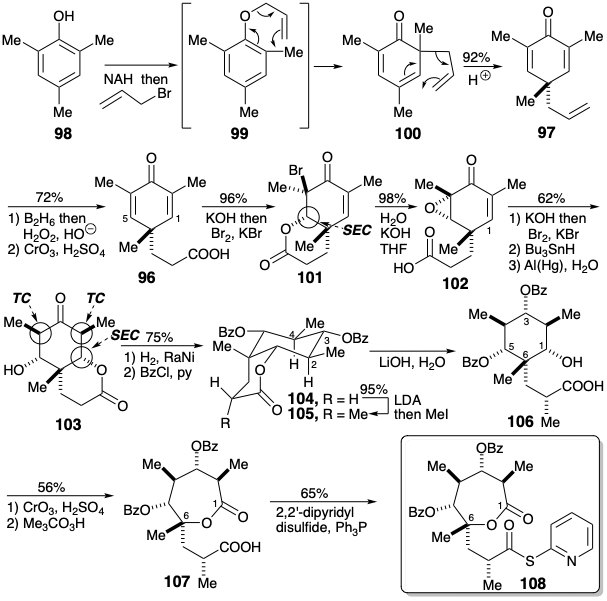
Нуклеофільна активація, що надається карбонілом лактону в 103, тепер може бути використана для введення метильної групи на позиції 8. Однак попереднє регулювання рівня функціональності в положенні 3 дозволяє уникнути нуклеофільної активації поруч з кетоном карбонілу. Метилювання 104 тоді дозволило 105 стереовибірково. Лактон в 103 диференціює гідроксильні замінники в положеннях 1 і 5. Щоб зберегти цю диференціацію після омилення лактону, інші гідроксили в 105 повинні були бути належним чином маскуються. Вибір групи маскування ефіру бензоату особливо тонкий. Доцільність селективного омилення лактону в присутності ефірів бензоату спирається на зменшену електрофільність бензоатної карбонільної групи внаслідок кон'югації. Окислення спирту 106 до кетону, а потім до лактону 107 з подальшою активацією карбонової кислоти у вигляді тіоефіру забезпечується електрофільний сегмент С1-9, еквівалентний 90, де маскують групи R 1 і R 4 замінюються лактоном. міст.
Синтез, описаний вище, забезпечує рацемічні сполуки. Дозвіл раннього проміжного продукту карбонової кислоти 102 шляхом фракційної кристалізації діастереомерних солей 1-α-нафтилетиламіну використовували для отримання проміжних продуктів з показаними вище абсолютними конфігураціями, які необхідні для природного еритроноліду В.
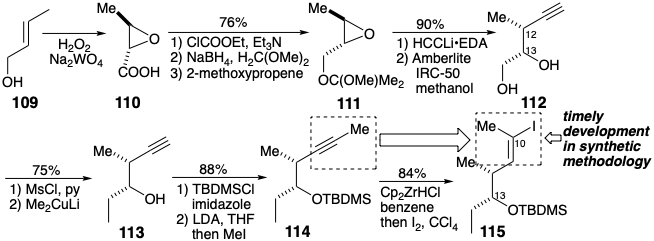
Дозвіл раннього проміжного також використовувався для підготовки необхідного енантіомера попередника 115 (див. Розділ 6.3) для нуклеофіла 89. Так, епоксидна карбонова кислота 110, яка легко доступна шляхом одностадійного окислення транс-кротилового спирту (109), була дозволена шляхом фракційної кристалізації діастереомерних 1-α-нафтилетиламінових солей. Стереоспецифічна нуклеофільна заміщення на 110 породила абсолютну конфігурацію, необхідну в позиції 12. Регіоселективність цього епоксидного отвору контролюється об'ємним ефірним замісником у 111. Використання відповідного епоксидного спирту при витісненні показало значно нижчу регіоселективність. Заміна термінального гідроксилу в 112 метильною групою, щоб дати 113, може бути здійснена без маскування вторинного гідроксилу за допомогою надлишку\(\ce{Me2CuLi}\). Повністю регіоселективне перетворення ацетилену 114 в йодид вінілу 115 залежало від надзвичайно високої регіоселективності, про яку нещодавно повідомлялося про гідроцирконування несиметрично двозаміщених ацетиленів. Цей крок синтезу еритроноліду В Корі є яскравим прикладом впливу розробок синтетичної методології на нашу здатність досягати ефективного синтезу складних органічних молекул.
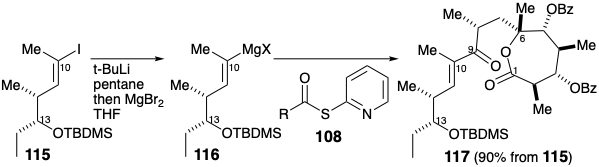
Хоча обидва будівельні блоки 108 і 115 були доступні в гомохіральній формі з правильними абсолютними конфігураціями для еритроноліду B, синтез фактично здійснювався шляхом з'єднання правильного енантіомера 115 з рацемічним 108. Таким чином, реагент Гріньяра 116, отриманий з 115, був ацильований тіоефіром 108 для отримання кетону 117 та діастереомера в 90% загального виходу. Суміш проводили через кілька додаткових етапів перед розділенням методом препаративної тонкошарової хроматографії.
Таким чином, зниження кетону карбонілу в 117 виявилося несподівано важким через дивно подібну реакційну здатність кето і лактонових карбонілів до більшості відновників, а також через схильність до кон'югатного скорочення енону. Зменшення борогідридом цинку супроводжувалося двома несподіваними явищами, дуже вітається повною стереоселективністю і по суті нерелевантною транслактонізацією, яка породила 10-членний лактон 118 після видалення силілозахисної групи в положенні 13. Омилення цього лактону було найбільш ефективно здійснено з\(\ce{LiOH}\) і водним,\(\ce{H2O2}\) який, імовірно, виграє від супернуклеофільності гідропероксиду аніону. Гідроліз менш реактивних ефірів бензоату в 119 був потім здійснений водним\(\ce{KOH}\). Подальша метил- нація доставляється 120 разом з діастереомером, з якого він був відокремлений ТЛК на силікагелі.
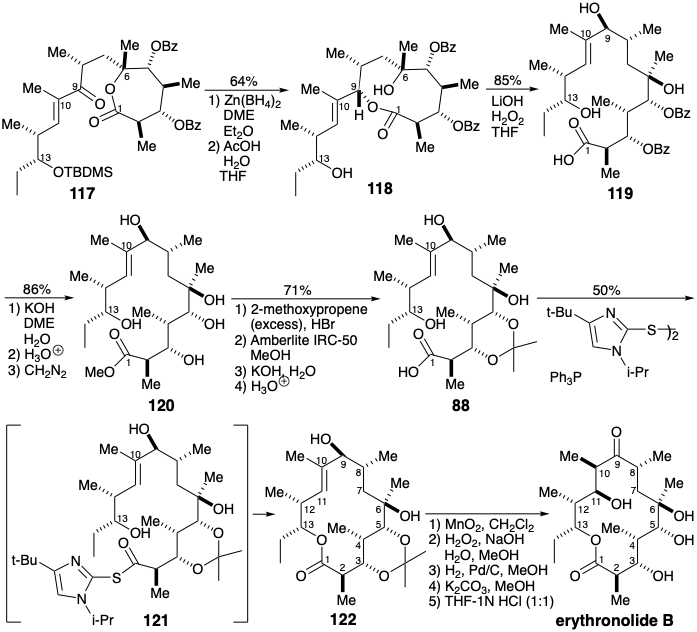
Релейна сполука 88 була отримана з 120 шляхом кеталізації 2-метоксипропеном, селективного гідролізу 2-метокси-2-пропілових ефірів, які також утворилися, і омилення метилового ефіру. Макролактонізація здійснювалася методом «подвійної активації», який передбачає одночасну активацію гідроксильної та карбоксильної функцій. Імовірно, подвійно активований проміжний 123 руйнується до тетраедричного карбонільного аддукту 124, з якого утворюється лактон 126 шляхом елімінації 125. Таким чином, нагрівання тіоефіру 122 при рефлюксі в сухому толуолі забезпечувало вихід еритрололіду В в 50%.
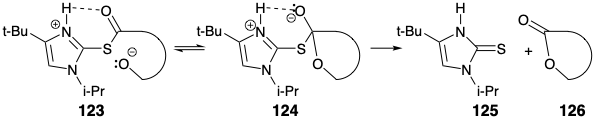
Еритронолід B з цукрових гомохіральних будівельних блоків.
Стратегія енантіоспецифічного загального синтезу еритроноліду В розвинулася з визнання того, що сегменти C2-4 та C10-12 ідентично заміщені, але мають різні абсолютні стереохімії. Такі сегменти, диференційовано замінені на кожному кінці, тобто 127 та 128, можуть бути розроблені та об'єднані для отримання натурального продукту. Тому були розпочаті дослідження з визначення синтетичних шляхів до таких проміжних продуктів. Оскільки (R) -2,3-о-ізопропіліденгліцеральдегід (129) є легкодоступним гомохіральним будівельним блоком (див. Розділ 3.7), було досліджено можливу корисність як вихідного матеріалу для енантіоспецифічного синтезу таких сегментів. 14
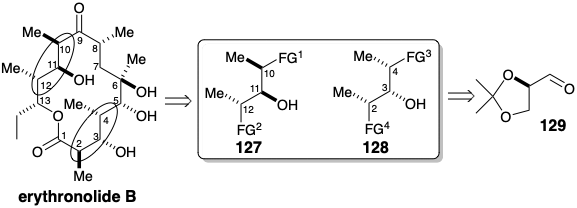
Додавання кротилхромового реагенту до альдегіду 129 практично не показало діастереофаціальної селективності для додавання до альдегіду, але високу перевагу генеруванню анти відносин у двох новоутворених стереоцентрах через стереоелектронну перевагу кріслоподібного перехідного стану. структури 130 і 131, які ведуть до 132 і 133. Ці діастереомери були легко відокремлені за допомогою препаративної колоночної хроматографії у великих масштабах. Перетворення 132 в проміжний тип 127 і 133 в інтермедіат типу 128 вимагає інверсії вільного вторинного гідроксилу і заміщення іншого вторинного кисневого замінника метилом з інверсією конфігурації. Інверсія вільного гідроксилу була здійснена шляхом активації у вигляді тозилату з подальшим внутрішньомолекулярним зміщенням S N 2 віцинальним гідроксилом. Цей стереоспеціально виробляв 135 з 134 і 138 з 137. Реакція цих епоксидів з\(\ce{Li2Me2CuCN}\) виконаною другою конфігураційною інверсією при заміні кисневого замінника метилом. Діоли 136 і 139 відповідають фрагментам 127 і 128 відповідно, де FG 1 і FG 3 є прихованими альдегідами, тоді як FG 2 і FG 4 є гідроксиметильними групами.
Наявність гомохіральних будівельних блоків 136 і 139 спрямувало другу фазу стратегічного планування. 15 Полярне відключення ерітроноліду B на C6-C7 і C8-C9 генерує два попередники, 140 і 142, обидва з термінальними карбонільними функціями. Полярне об'єднання цих фрагментів вимагало б «віцинально діаніонного двовуглецевого (C6/C7) синтону» 141 з підвісною метильною групою. Хоча ідентифікація синтетичного еквівалента для 141 була відкладена, було визнано, що «метильне розгалуження виключає пряме застосування деякого ацетиленового похідного».
Подальше полярне відключення 142 для створення альдегіду 143, який повинен бути доступний з будівельного блоку 139, вимагає ацетилкарбоніонного синтону, для якого ізопропенілмагнію бромід є прихованим синтетичним еквівалентом. Алкен 144 є прихованим еквівалентом альдегіду 140. Подальша полярна дислокація 144 передбачає етиловий нуклеофіл та альдегід електрофіл 145, який повинен бути доступний шляхом окислення 136.
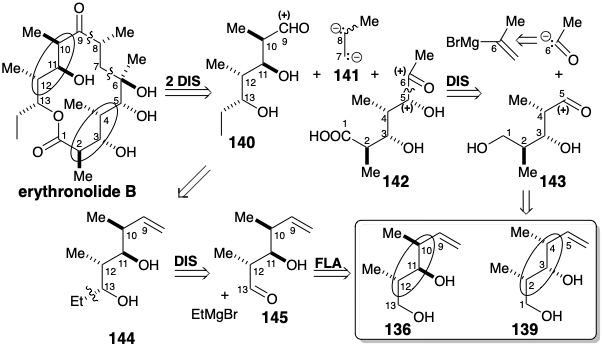
Масковане похідне 147 з 142, що містить функціональність карбонової кислоти в прихованій формі у вигляді бензилоксиефіру, готували з гомохірального будівельного блоку 139 (див. нижче). Додавання реагенту Гріньяра до проміжного альдегіду породило стереоцентр у позиції 5 нестереоселективно, що призводить до 146 у вигляді суміші діастереомерів 2:1. Однак необхідна конфігурація на цьому вуглеці може бути встановлена шляхом врівноваження епімерних кетонів 147 і 148. Екваторіальний кетон був прихильний над осьовим епімером 148 на 94:6 при рівновазі.
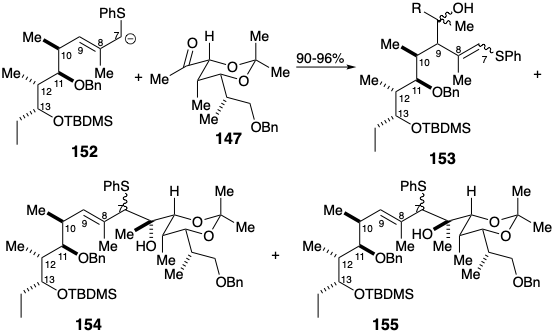
Масковане похідне 149 альдегіду 140 було отримано з гомохірального будівельного блоку 136. Хоча полярне об'єднання двох карбонілів, що містять фрагменти 147 і 149, може використовувати дисонансний діаніонний фрагмент, відповідний 141, синтетичний еквівалент 141 не був розроблений. Швидше за все, нуклеофільний реагент, який був нуклеофільним при С6 і містив електрофільний вуглець у положенні 7, був з'єднаний з альдегідом 151, а потім полярна реакційна здатність C7 була інвертована шляхом перетворення в алліловий тіоефір, який може бути депротонований для забезпечення нуклеофільної реактивності при C7.

Вуглецевий скелет ерітроноліду В був завершений з'єднанням кетону 147 з алліловим карбоніоном 152, отриманим шляхом депротонації сульфіду 151 з N-булі в присутності TMEDA. Початкові результати були розчаровуючими, оскільки основним продуктом був γ-аддукт 153, а не бажаний α-аддукт 154. За рахунок додавання HMPA утворення 153 можна було придушити майже повністю. Однак в цих умовах основним продуктом був епімерний α-аддукт 155. Нарешті, було виявлено, що прекомплексоутворення кетону 147 з\(\ce{BF3}\) сильно сприяє необхідної області і стереоселективності.
Служивши своїй меті оператором інверсії полярної реактивності, алліловий фенілтіозамісник видаляли редуктивно. Потім диференціація гідроксильних груп була здійснена ацетилюванням з подальшим селективним деацетилюванням 156 для розкриття первинного гідроксилу. Окислення з подальшим вичерпним деацетилюванням доставляється тригідроксикислота 157. Макролактонізація була accomp- lished в дуже хорошому врожайності шляхом перетворення в змішаний ангідрид, який циклізувався в розведеному розчині толуолу. Мабуть, конформація без деформації, яка ідеально підходить для циклізації, доступна до 157, тоді як серйозні затори присутні в конформаціях, придатних для формування 12-членного лактону шляхом ацилювання 11-OH.
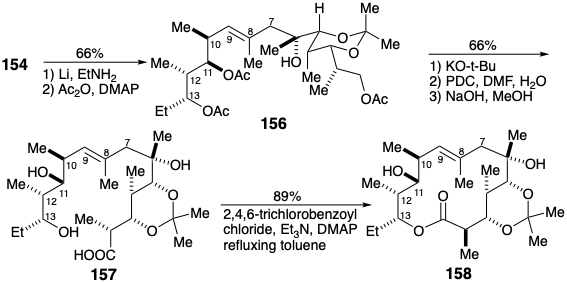
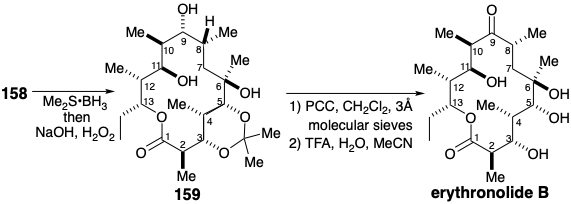
Завершення синтезу вимагало введення кисню в положення 9. Антимарковнікова гідратація зв'язку 8,9-C=C у 158 шляхом гідроборації-окислення здійснила цю функціоналізацію, і, очевидно, завдяки макроциклічним конформаційним ефектам генерації правильної конфігурації в положенні 8 сприяло 9:1. Конформаційні ефекти також сприяли селективному окисленню вторинного гідроксилу в положенні 9 у присутності іншого вторинного гідроксилу в положенні 11. Таким чином, накопичення багатьох сприятливо вибіркових кроків внаслідок тонких, непередбачуваних наслідків молекулярної форми - тобто надзвичайно ефективної макролактонізації та сприятливо стерео- та регіоселективних процесів - призвело до тотального синтезу, який конкурує зі стратегією Корі, яка була більшою ретельно спланований шляхом ретельного ретеросинтетичного аналізу.