11.3: Кулометричні методи
- Page ID
- 24952

У потенціометричному методі аналізу визначають концентрацію аналіта шляхом вимірювання потенціалу електрохімічної клітини в статичних умовах, в яких струм не протікає, а концентрації видів в електрохімічній комірці залишаються фіксованими. Динамічні методи, при яких струм проходить через електрохімічну комірку і змінюються концентрації, також є важливими електрохімічними методами аналізу. У цьому розділі ми розглянемо кулометрію. Вольтамметрія і амперометрія розглянуті в розділі 11.4.
Кулометрія заснована на вичерпному електролізі аналіта. Під вичерпним ми маємо на увазі, що аналіт окислюється або повністю відновлюється на робочому електроді, або що він повністю реагує з реагентом, що утворюється на робочому електроді. Існує дві форми кулометрії: кулометрія з контрольованим потенціалом, при якій ми застосовуємо постійний потенціал до електрохімічної осередку, і кулометрія контрольованого струму, при якій ми пропускаємо постійний струм через електрохімічну комірку.
Під час електролізу загальний заряд Q в кулоні, який проходить через електрохімічну комірку, пропорційний абсолютній кількості аналіту за законом Фарадея
де n - кількість електронів на моль аналіту, F - константа Фарадея (96 487 C моль —1), а N A - молі аналіту. Кулон еквівалентний a•сек; таким чином, для постійного струму i сумарний заряд дорівнює
де t e - час електролізу. Якщо струм змінюється з часом, як це відбувається в кулометрії з контрольованим потенціалом, то сумарний заряд дорівнює
\[Q=\int_{0}^{t_e} i(t) d t \label{11.3}\]
У кулометрії ми відстежуємо струм як функцію часу і використовуємо Equation\ ref {11.2} або Equation\ ref {11.3} для обчислення Q. Знаючи загальний заряд, ми потім використовуємо Equation\ ref {11.1} для визначення молей аналіту. Для отримання точного значення для N А весь струм повинен окислювати або зменшувати аналіт; тобто кулометрія вимагає 100% ККД струму або точного вимірювання ККД струму за допомогою стандарту.
ККД струму - це відсоток струму, який фактично призводить до окислення або відновлення аналіта.
Кулометрія з керованим потенціалом
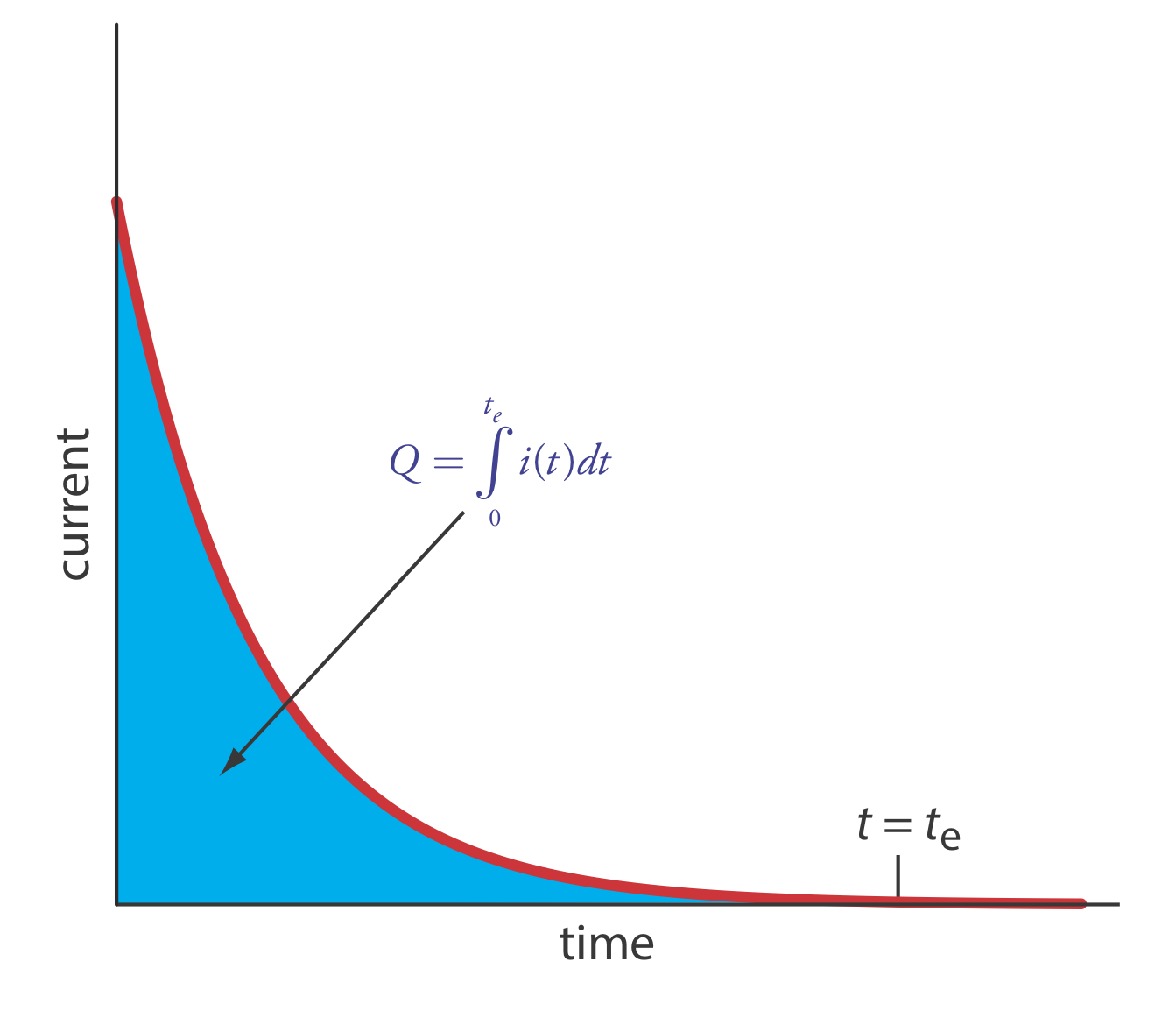
Найпростіший спосіб забезпечити 100% ефективність струму - утримувати робочий електрод при постійному потенціалі, коли аналіт окислюється або відновлюється повністю і де не окислюються або відновлюються будь-які потенційні заважаючі види. У міру прогресування електролізу концентрація аналіта і струм зменшуються. Отриманий профіль струму проти часу для кулометрії з керованим потенціалом показано на рисунку Template:index. Інтеграція площі під кривою (Equation\ ref {11.3}) від t = 0 до t = t e дає загальний заряд. У цьому розділі розглядаються експериментальні параметри та приладобудування, необхідні для розробки кулометричного методу аналізу з керованим потенціалом.
Вибір постійного потенціалу
Щоб зрозуміти, як підбирається відповідний потенціал для робочого електрода, розробимо кулометричний метод з постійним потенціалом для Cu 2+, заснований на його відновленні до мідного металу на робочому електроді Pt.
\[\mathrm{Cu}^{2+}(a q)+2 e^{-} \rightleftharpoons \mathrm{Cu}(s) \label{11.4}\]
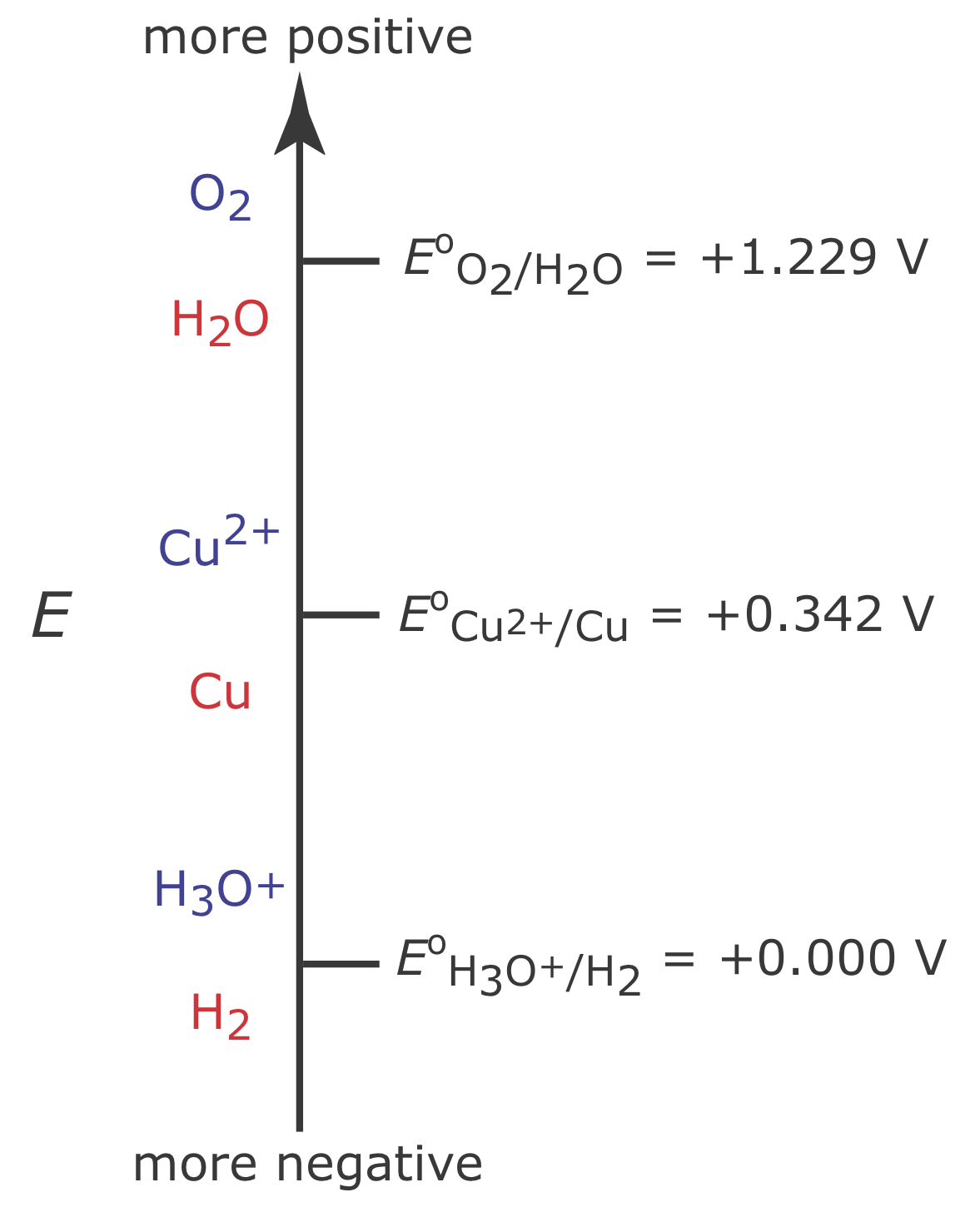
На малюнку Template:index показана сходова діаграма для водного розчину Cu 2 +. З сходової діаграми ми знаємо, що реакція\ ref {11.4} сприятлива, коли потенціал робочого електрода більш негативний, ніж +0,342 В проти стандартного водневого електрода. Однак для забезпечення 100% ефективності струму потенціал повинен бути досить позитивним, ніж +0.000 В, щоб зниження Н 3 О + до Н 2 не сприяло суттєво сумарному струму, що протікає через електрохімічну комірку.
Ми можемо використовувати рівняння Нернста для реакції\ ref {11.4} для оцінки мінімального потенціалу кількісного зменшення Cu 2 +.
Так чому ж ми використовуємо концентрацію Cu 2 + в Equation\ ref {11.5} замість його активності? У потенціометрії ми використовуємо активність, оскільки ми використовуємо E клітину для визначення концентрації аналіта. Тут ми використовуємо рівняння Нернста, щоб допомогти нам вибрати відповідний потенціал. Як тільки ми виявимо потенціал, ми можемо скорегувати його значення в міру необхідності, щоб забезпечити кількісне зниження Cu 2 +. Крім того, в кулометрії концентрація аналіта задається загальним зарядом, а не прикладеним потенціалом.
Якщо визначити кількісний електроліз як такий, при якому ми зменшуємо 99,99% Cu2+ до Cu, то концентрація Cu 2+ при t e дорівнює
де [Cu 2 +] 0 - початкова концентрація Cu 2 + в зразку. Підставляючи рівняння\ ref {11.6} в рівняння\ ref {11.5}, ми можемо обчислити необхідний потенціал.
\[E=E_{\mathrm{Cu}^{2+} / \mathrm{Cu}}^{\circ}-\frac{0.05916}{2} \log \frac{1}{0.0001 \times\left[\mathrm{Cu}^{2+}\right]} \nonumber\]
Якщо початкова концентрація Cu 2+ дорівнює\(1.00 \times 10^{-4}\) M, то потенціал робочого електрода повинен бути більше негативного, ніж +0,105 В, щоб кількісно зменшити Cu 2 + до Cu. Відзначимо, що при цьому потенціал Н 3 О + не знижується до Н 2, зберігаючи 100% коефіцієнт корисної дії струму.
Багато кулометричні методи з контрольованим потенціалом для Cu 2 + використовують потенціал, який є негативним відносно стандартного електрода водню - див., наприклад, Рехніц, Г.А. Аналіз контрольованого потенціалу, Макміллан: Нью-Йорк, 1963, с.49. Виходячи з діаграми сходів на малюнку Template:index, ви можете очікувати, що застосування потенціалу <0.000 V частково зменшить H 3 O + до H 2, що призведе до струму ККД менше 100%. Причина, по якій ми можемо використовувати такий негативний потенціал, полягає в тому, що швидкість реакції на зниження Н 3 О + до Н 2 дуже повільна у електрода Pt. Це призводить до значного перенапруження - необхідності застосування потенціалу, більш позитивного або більш негативного, ніж передбачене термодинамікою, - що зміщує E o для окислювально-відновлювальної пари H 3 O +/H 2 до більш негативної. значення.
Мінімізація часу електролізу
У кулометрії з контрольованим потенціалом, як показано на рисунку Template:index, струм зменшується з часом. Як результат, швидкість електролізу - нагадаємо з глави 11.1, що струм є мірою швидкості - стає повільнішою, і вичерпний електроліз аналіту може зажадати тривалого часу. Оскільки час є важливим фактором при розробці аналітичного методу, ми повинні враховувати фактори, які впливають на час аналізу.
Ми можемо наблизити зміну струму як функцію часу на рисунку Template:index як експоненціальний спад; таким чином, струм часу t дорівнює
\[i_{t}=i_{0} e^{-k t} \label{11.7}\]
де i 0 - струм при t = 0 і k - постійна швидкості, яка прямо пропорційна площі робочого електрода і швидкості перемішування, і що обернено пропорційно обсягу розчину. Для вичерпного електролізу, при якому ми окислюємо або зменшуємо 99,99% аналіту, струм в кінці аналізу, т е, дорівнює
\[i_{t_{e}} \leq 0.0001 \times i_{0} \label{11.8}\]
Підставляючи рівняння\ ref {11.8} на рівняння\ ref {11.7} і розв'язування для t e дає мінімальний час для вичерпного електролізу як
\[t_{e}=-\frac{1}{k} \times \ln (0.0001)=\frac{9.21}{k} \nonumber\]
З цього рівняння ми бачимо, що більша величина для k скорочує час аналізу. З цієї причини ми зазвичай проводимо кулометричний аналіз з контрольованим потенціалом в електрохімічній комірці невеликого об'єму, використовуючи електрод з великою площею поверхні та з високою швидкістю перемішування. Кількісний електроліз зазвичай вимагає приблизно 30-60 хв, хоча можливі коротші або тривалі терміни.
Контрольно-вимірювальні прилади
Триелектродний потенціостат використовується для установки потенціалу в кулометрії з керованим потенціалом (див. Рис. Робочі електроди зазвичай бувають двох типів: циліндричний електрод Pt, виготовлений з платинової марлі (рис. Template:index), або електрод Hg басейну. Великий перенапруга для зниження H 3 O + при Hg робить його електродом вибору для аналіту, який вимагає негативного потенціалу. Наприклад, потенціал, більш негативний, ніж —1 В проти SHE, можливий при електроді Hg, але не на електроді Pt - навіть у дуже кислому розчині. Оскільки ртуть легко окислюється, вона менш корисна, якщо нам потрібно підтримувати потенціал, який є позитивним щодо SHE. Платина - це робочий електрод вибору, коли нам потрібно застосувати позитивний потенціал.

Допоміжний електрод, який часто є проводом Pt, відокремлений соляним містком від аналітичного розчину. Це необхідно для того, щоб продукти електролізу, що утворюються на допоміжному електроді, не вступали в реакцію з аналітом і не втручалися в аналіз. В якості опорного електрода служить насичений каломель або Ag/AgCl електрод.
Інша істотна потреба в кулометрії з контрольованим потенціалом - це засіб для визначення сумарного заряду. Одним із методів є моніторинг струму як функція часу та визначення площі під кривою, як показано на малюнку Template:index. Сучасні прилади використовують електронну інтеграцію для контролю заряду як функції часу. Загальний заряд в кінці електролізу зчитується безпосередньо з цифрового зчитування.
Електрогравіметрія
Якщо твір кулометрії з керованим потенціалом утворює наліт на робочому електроді, то в якості аналітичного сигналу можна використовувати зміну маси електрода. Наприклад, якщо застосувати потенціал, який зменшує Cu 2 + до Cu на робочому електроді Pt, різниця в масі електрода до і після електролізу є прямим вимірюванням кількості міді в зразку. Як ми дізналися в розділі 8, ми називаємо аналітичну техніку, яка використовує масу як сигнал, гравіметричною технікою; таким чином, ми називаємо цю електрогравіметрію.
Кулометрія контрольованого струму
Другий підхід до кулометрії полягає у використанні постійного струму замість постійного потенціалу, що призводить до формування профілю струму проти часу, показаного на рисунку Template:index. Кулометрія контрольованого струму має дві переваги перед кулометрією з керованим потенціалом. По-перше, час аналізу коротший, оскільки струм не зменшується з часом. Типовий час аналізу кулометрії з керованим струмом менше 10 хв, у порівнянні з приблизно 30-60 хв для кулометрії з керованим потенціалом. По-друге, оскільки загальний заряд просто є добутком струму та часу (Equation\ ref {11.2}), немає необхідності інтегрувати криву поточного часу на рис. Template:index.
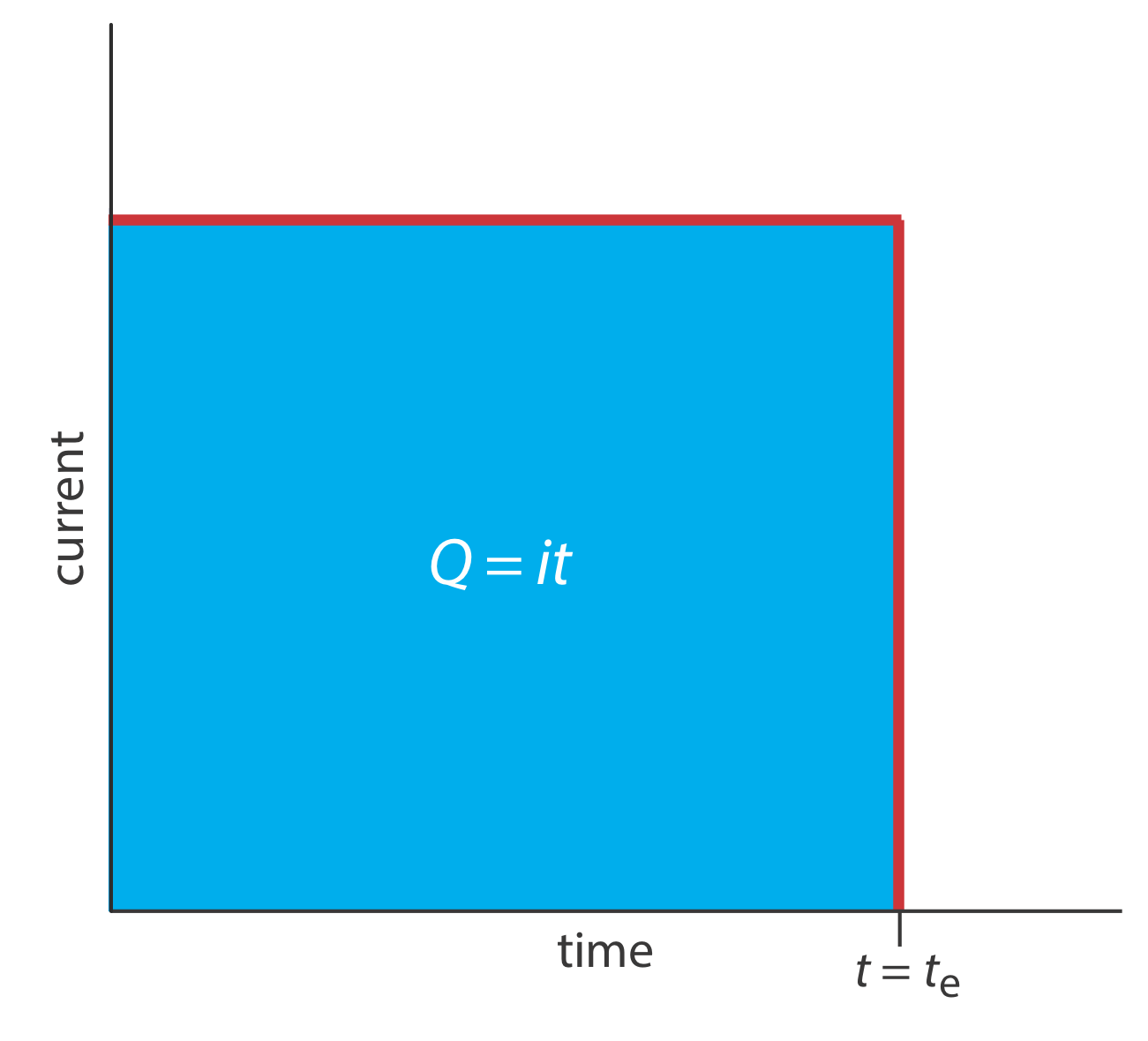
Використання постійного струму представляє нам дві важливі експериментальні проблеми. По-перше, під час електролізу концентрація аналіту - і, отже, струм, що виникає в результаті його окислення або відновлення, постійно зменшується. Для підтримки постійного струму ми повинні дозволити потенціалу змінюватися до тих пір, поки на робочому електроді не відбудеться інша реакція окислення або відновлення. Якщо ми не спроектуємо систему ретельно, ця вторинна реакція призводить до ефективності струму, який менше 100%. Друга проблема полягає в тому, що нам потрібен метод, щоб визначити, коли електроліз аналіта завершений. Як показано на малюнку Template:index, в кулометричному аналізі з контрольованим потенціалом ми знаємо, що електроліз завершується, коли струм досягає нуля, або коли він досягає постійного фону або залишкового струму. Однак при кулометричному аналізі з контрольованим струмом струм продовжує протікати навіть тоді, коли електроліз аналіта завершений. Необхідний відповідний метод визначення кінцевої точки реакції, т е.
Підтримка поточної ефективності
Щоб проілюструвати, чому зміна потенціалу робочого електрода може привести до ККД струму менше 100%, розглянемо кулометричний аналіз для Fe 2 + на основі його окислення до Fe 3 + при робочому електроді Pt в 1 M H 2 SO 4.
\[\mathrm{Fe}^{2+}(a q) \rightleftharpoons \text{ Fe}^{3+}(a q)+e^{-} \nonumber\]
На малюнку Template:index показана діаграма сходів для цієї системи. На початку аналізу потенціал робочого електрода залишається майже постійним на рівні, близькому до його початкового значення.
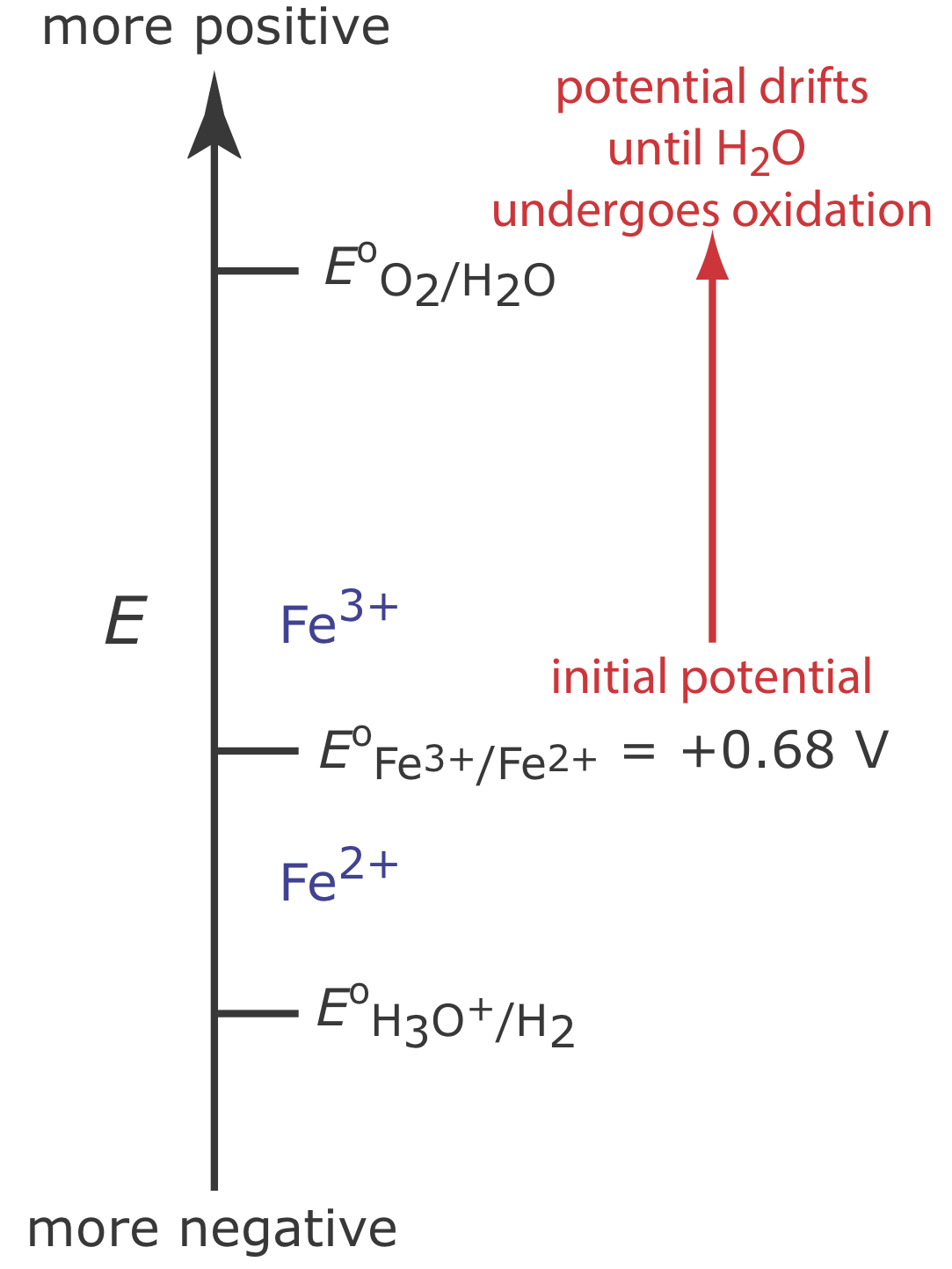
У міру зменшення концентрації Fe 2 + і збільшення концентрації Fe 3 + потенціал робочого електрода зміщується в бік більш позитивних значень, поки не почнеться окислення Н 2 О.
\[2 \mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons \text{ O}_{2}(g)+4 \mathrm{H}^{+}(a q)+4 e^{-} \nonumber\]
Оскільки частина загального струму надходить від окислення H 2 O, ефективність струму для аналізу менше 100%, і ми не можемо використовувати Equation\ ref {11.1} для визначення кількості Fe 2 + у зразку.
Хоча ми не можемо запобігти дрейфу потенціалу, поки інший вид не піддається окисленню, ми можемо підтримувати 100% ефективність струму, якщо продукт цієї реакції вторинного окислення як швидко, так і кількісно реагує з рештою Fe 2 +. Для цього додаємо надлишок Ce 3 + до аналітичного рішення. Як показано на малюнку Template:index, коли потенціал робочого електрода зміщується до більш позитивного потенціалу, Ce 3 + починає окислюватися до Ce 4 +
\[\mathrm{Ce}^{3+}(a q) \rightleftharpoons \text{ Ce}^{4+}(a q)+e^{-} \label{11.9}\]
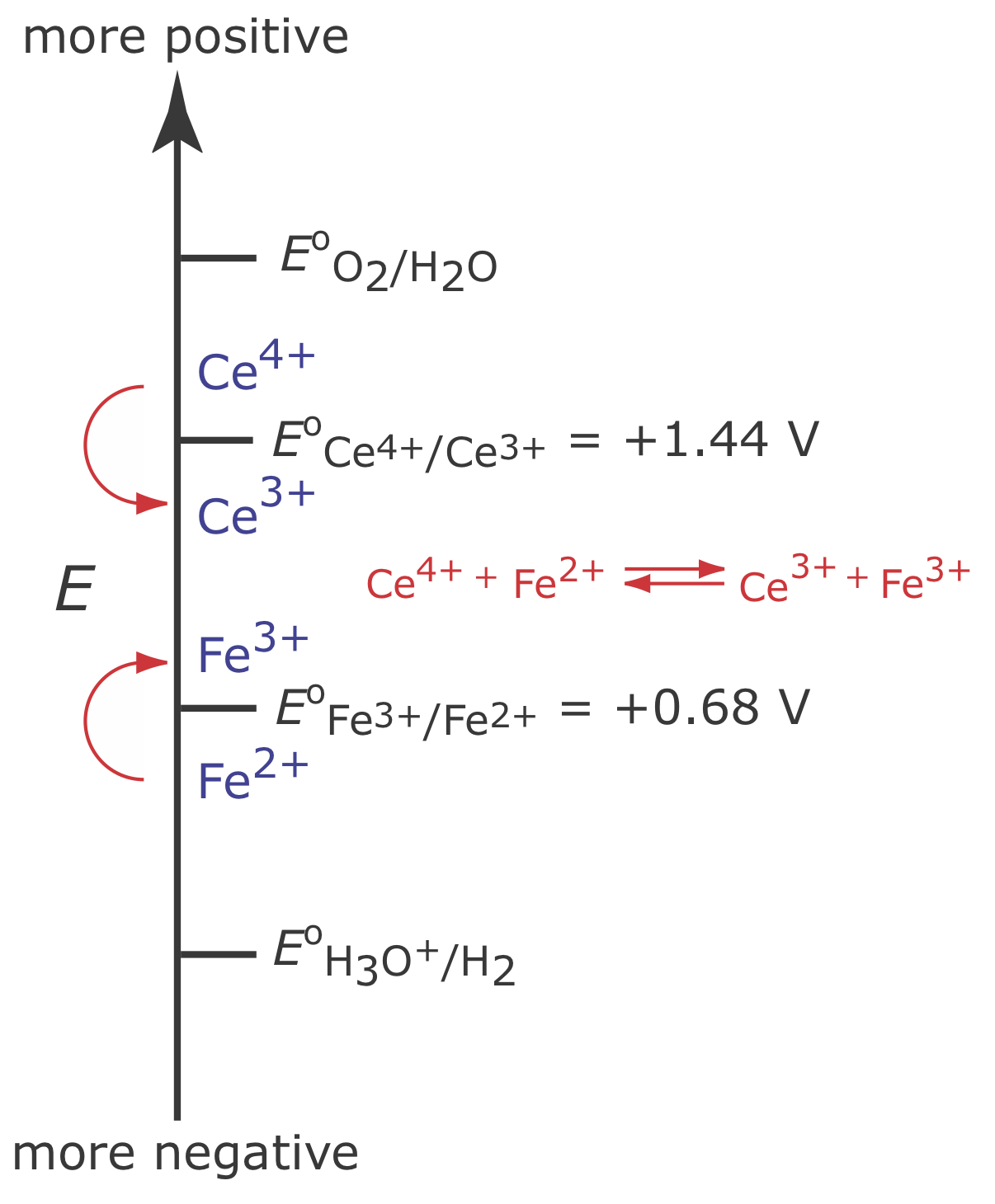
Се 4 +, що утворюється на робочому електроді, швидко змішується з розчином, де вступає в реакцію з будь-яким наявним Fe 2 +.
Поєднання реакції\ ref {11.9} і реакції\ ref {11.10} показує, що чистою реакцією є окислення Fe 2 + до Fe 3 +
\[\mathrm{Fe}^{2+}(a q) \rightleftharpoons \text{ Fe}^{3+}(a q)+e^{-} \nonumber\]
який підтримує коефіцієнт корисної дії струму 100%. Вид, який використовується для підтримки 100% ефективності струму, називається медіатором.
Визначення кінцевої точки
Додавання медіатора вирішує проблему збереження 100% ефективності струму, але це не вирішує завдання визначення того, коли електроліз аналіта завершений. Використовуючи аналіз для Fe 2 + на рис. Template:index, коли окислення Fe 2 + закінчується повний струм продовжує витікати від окислення Ce 3 +, і, врешті-решт, окислення H 2 O. Що нам потрібно, це сигнал, який повідомляє нам, коли в розчині більше немає Fe 2 +.
Для наших цілей кулометричний аналіз з контрольованим струмом зручно розглядати як реакцію між аналітом, Fe 2 +, і медіатором Ce 3 +, як показано реакцією\ ref {11.10}. Ця реакція ідентична окислювально-відновному титруванню; таким чином, ми можемо використовувати кінцеві точки для окислювально-відновного титрування - візуальних показників та потенціометричних або кондуктометричних вимірювань - щоб сигналізувати про закінчення кулометричного аналізу контрольованого струму. Наприклад, фероін забезпечує корисну візуальну кінцеву точку для опосередкованого кулометричного аналізу Ce 3 + для Fe 2 +, змінюючи колір з червоного на синій при завершенні електролізу Fe 2 +.
Реакція\ ref {11.10} - це та сама реакція, яку ми використовували в Главі 9 для розвитку нашого розуміння окислювально-відновної титриметрії.
Контрольно-вимірювальні прилади
Керовано-струмова кулометрія в нормі здійснюється за допомогою двоелектродного гальваностата, який складається з робочого електрода і зустрічного електрода. Робочий електрод - часто простий електрод Pt - також називають електродом генератора, оскільки саме там медіатор реагує на генерацію виду, який реагує з аналітом. При необхідності зустрічний електрод ізолюють від аналітичного розчину сольовим містком або пористою фриттою, щоб запобігти реакції його продуктів електролізу з аналітом. Крім того, ми можемо генерувати окислювач або відновник зовні і дозволити йому текти в аналітичний розчин. На малюнку Template:index показано один простий метод для цього. Розчин, що містить медіатор, перетікає в електрохімічну комірку невеликого об'єму, продукти виходять через окремі трубки. Залежно від аналіту окислювач або відновлювальний реагент доставляється в аналітичний розчин. Наприклад, ми можемо генерувати Ce 4 + за допомогою водного розчину Ce 3 +, направляючи Ce 4 +, який утворюється на аноді, до нашого зразка.
На малюнку 11.1.4 показаний приклад ручного гальваностата. Хоча сучасний гальваностат використовує дуже різні схеми, ви можете використовувати рис. 11.1.4 та супровідну дискусію, щоб зрозуміти, як ми можемо використовувати робочий електрод і зустрічний електрод для управління струмом. Малюнок 11.1.4 включає додатковий опорний електрод, але його наявність або відсутність не важливо, якщо ми не зацікавлені в контролі потенціалу робочого електрода.
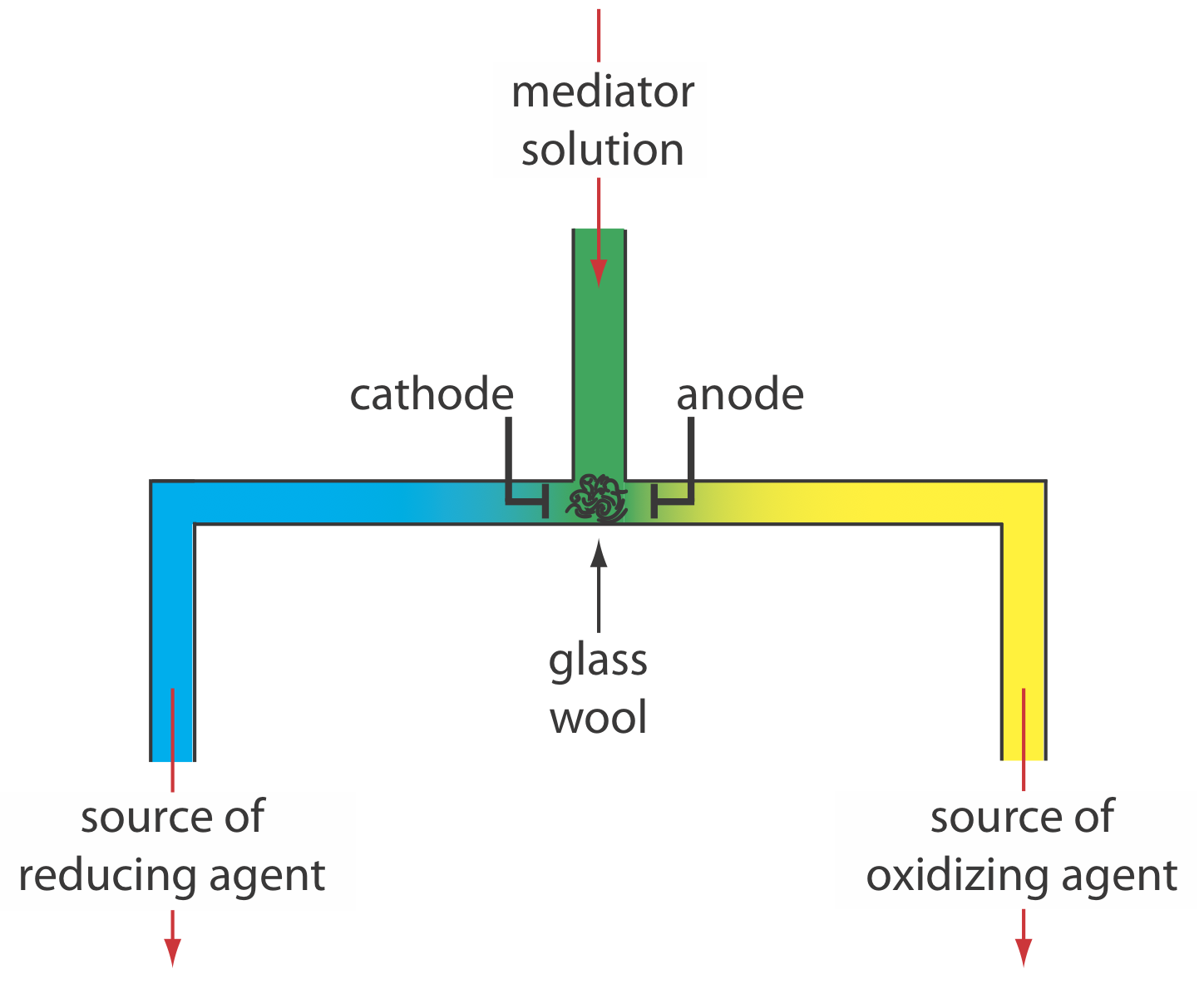
Є ще дві найважливіші потреби в кулометрії контрольованого струму: точний годинник для вимірювання часу електролізу, т е, і перемикач для запуску і зупинки електролізу. Аналоговий годинник може записувати час до найближчого ± 0,01 с, але необхідність зупинки та запуску електролізу при наближенні до кінцевої точки може призвести до загальної невизначеності ± 0,1 с Цифровий годинник дозволяє більш точно виміряти час із загальною невизначеністю ± 1 мс. Перемикач повинен контролювати як струм, так і годинник, щоб ми могли зробити точне визначення часу електролізу.
Кулометричні титрування
Кулометричний метод контрольованого струму іноді називають кулометричним титруванням через його схожість зі звичайним титруванням. Наприклад, в кулометричному аналізі контрольованого струму для Fe 2 + за допомогою медіатора Ce 3 + окислення Fe 2 + Ce 4 + (реакція\ ref {11.10}) ідентично реакції при окислювально-відновному титруванні.
Є й інші подібності між кулометрією з контрольованим струмом і титриметрією. Якщо об'єднати Equation\ ref {11.1} і Equation\ ref {11.2} і розв'яжемо для молів аналіту N A, то отримаємо наступне рівняння.
\[N_{A}=\frac{i}{n F} \times t_{e} \label{11.11}\]
Порівняйте рівняння\ ref {11.11} із співвідношенням між молями аналіту, N A та молями титранту, N T, у титруванні
\[N_{A}=N_{T}=M_{T} \times V_{T} \nonumber\]
де M T і V T - молярність титранта і об'єм титранту в кінцевій точці. У кулометрії з постійним струмом джерело струму еквівалентно титранту, і значення цього струму аналогічно молярності титранта. Час електролізу аналогічний об'єму титранту, а t e еквівалентно кінцевій точці титрування. Нарешті, перемикач пуску і зупинки електролізу виконує ту ж функцію, що і запірний кран бюрета.
Для простоти ми припустили вище, що стехіометрія між аналітом і титрантом дорівнює 1:1. Однак припущення не є важливим і не впливає на наше спостереження за подібністю між кулометрією з контрольованим струмом і титруванням.
Кількісні програми
Кулометрія використовується для кількісного аналізу як неорганічних, так і органічних аналітів. Приклади кулометричних методів з керованим потенціалом і керованим струмом розглядаються в наступних двох розділах.
Кулометрія з керованим потенціалом
Більшість кулометричних аналізів з контрольованим потенціалом передбачають визначення неорганічних катіонів та аніонів, включаючи мікрометали та іони галогенідів. Таблиця Template:index узагальнює декілька з цих методів.
| аналіт | електролітична реакція | електрод |
|---|---|---|
| сурму | \(\text{Sb}(\text{III}) + 3 e^{-} \rightleftharpoons \text{Sb}\) | Пт |
| миш'яку | \(\text{As}(\text{III}) \rightleftharpoons \text{As(V)} + 2 e^{-}\) | Пт |
| кадмію | \(\text{Cd(II)} + 2 e^{-} \rightleftharpoons \text{Cd}\) | Pt або Hg |
| кобальт | \(\text{Co(II)} + 2 e^{-} \rightleftharpoons \text{Co}\) | Pt або Hg |
| мідь | \(\text{Cu(II)} + 2 e^{-} \rightleftharpoons \text{Cu}\) | Pt або Hg |
| галогеніди (X —) | \(\text{Ag} + \text{X}^- \rightleftharpoons \text{AgX} + e^-\) | Ag |
| залізо | \(\text{Fe(II)} \rightleftharpoons \text{Fe(III)} + e^-\) | Пт |
| свинець | \(\text{Pb(II)} + 2 e^{-} \rightleftharpoons \text{Pb}\) | Pt або Hg |
| нікель | \(\text{Ni(II)} + 2 e^{-} \rightleftharpoons \text{Ni}\) | Pt або Hg |
| плутонію | \(\text{Pu(III)} \rightleftharpoons \text{Pu(IV)} + e^-\) | Пт |
| срібло | \(\text{Ag(I)} + 1 e^{-} \rightleftharpoons \text{Ag}\) | Пт |
| олово | \(\text{Sn(II)} + 2 e^{-} \rightleftharpoons \text{Sn}\) | Пт |
| уран | \(\text{U(VI)} + 2 e^{-} \rightleftharpoons \text{U(IV})\) | Pt або Hg |
| цинку | \(\text{Zn(II)} + 2 e^{-} \rightleftharpoons \text{Zn}\) | Pt або Hg |
|
Джерело: Rechnitz, Г.А. Аналіз контрольованого потенціалу, Макміллан: Нью-Йорк, 1963. Електролітичні реакції записуються з точки зору зміни ступеня окислення аналіта. Фактичний вид в розчині залежить від аналіту. |
||
Можливість контролювати селективність шляхом регулювання потенціалу робочого електрода робить кулометрію з керованим потенціалом особливо корисною для аналізу сплавів. Наприклад, ми можемо визначити склад сплаву, що містить Ag, Bi, Cd і Sb, розчиняючи зразок і помістивши його в матрицю 0,2 M H 2 SO 4 разом з робочим електродом Pt і зустрічним електродом Pt. Якщо застосувати постійний потенціал +0,40 В проти SCE, Ag (I) відкладення на електроді як Ag та інші іони металів залишаються в розчині. Коли електроліз завершений, ми використовуємо загальний заряд для визначення кількості срібла в сплаві. Далі зміщуємо потенціал робочого електрода на -0,08 В проти СКВ, відкладаючи Bi на робочий електрод. Коли кулометричний аналіз для вісмуту завершено, визначаємо сурму шляхом зміщення потенціалу робочого електрода до —0,33 В проти СКВ, осаджуючи Sb. Нарешті, ми визначаємо кадмій після його електроосадження на робочий електрод з потенціалом —0,80 В проти СКВ.
Ми також можемо використовувати кулометрію з контрольованим потенціалом для кількісного аналізу органічних сполук, хоча кількість застосувань значно менше, ніж для неорганічних аналітів. Одним із прикладів є шестиелектронне відновлення нітрогрупи, —NO 2, до первинного аміну, —NH 2, на ртутному електроді. Розчини пікринової кислоти - також відомий як 2,4,6-тринітрофенол, або TNP, близький родич ТНТ - аналізується шляхом зниження його до триамінофенолу.
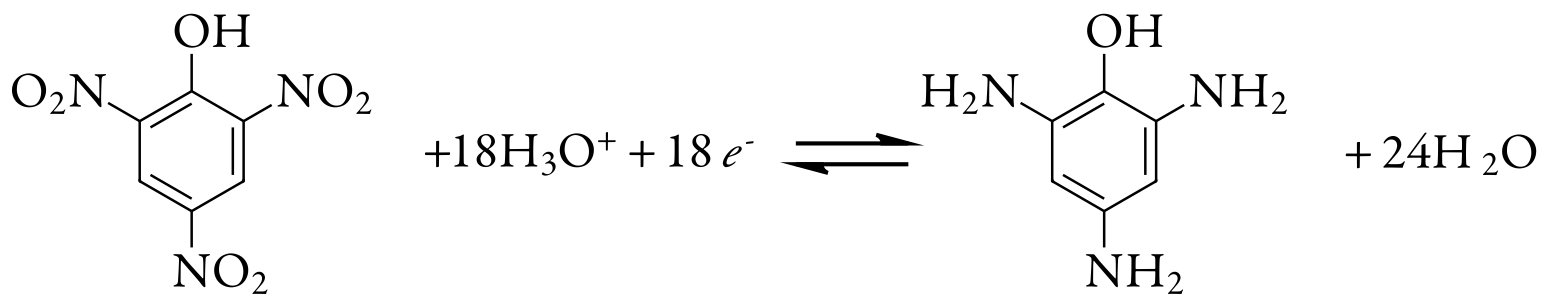
Іншим прикладом є послідовне зменшення трихлорацетату до дихлорацетату, а дихлорацетату - до монохлорацетату.
\[\text{Cl}_3\text{CCOO}^-(aq) + \text{H}_3\text{O}^+(aq) + 2 e^- \rightleftharpoons \text{Cl}_2\text{HCCOO}^-(aq) + \text{Cl}^-(aq) + \text{H}_2\text{O}(l) \nonumber\]
\[\text{Cl}_2\text{HCCOO}^-(aq) + \text{ H}_3\text{O}^+(aq) + 2 e^- \rightleftharpoons \text{ ClH}_2\text{CCOO}^-(aq) + \text{ Cl}^-(aq) + \text{H}_2\text{O}(l) \nonumber\]
Ми можемо проаналізувати суміш трихлорацетату та дихлорацетату, вибравши початковий потенціал, де реагує лише більш легко знижений трихлорацетат. Коли його електроліз завершиться, ми можемо зменшити дихлорацетат, регулюючи потенціал до більш негативного потенціалу. Загальний заряд для першого електролізу дає кількість трихлорацетата, а різниця в загальному заряді між першим електролізом і другим електролізом дає кількість дихлорацетата.
Кулометрія контрольованого струму (кулометричні титрування)
Використання медіатора робить кулометричне титрування більш універсальним аналітичним прийомом, ніж кулометрія з керованим потенціалом. Наприклад, пряме окислення або відновлення білка на робочому електроді важко, якщо активний окислювально-відновний ділянку білка лежить глибоко в його структурі. Кулометричне титрування білка можливо, однак, якщо ми використовуємо окислення або відновлення медіатора для отримання виду розчину, який реагує з білком. Таблиця Template:index узагальнює декілька кулометричних методів контрольованого струму, заснованих на окислювально-відновній реакції з використанням медіатора.
| посередник | електрохімічно генерований реагент і реакція | представник додаток |
|---|---|---|
| Вік + | \(\mathrm{Ag}^{+} \rightleftharpoons \textbf{Ag}^\textbf{2+}+e^{-}\) | \ (\ mathbf {H} _ {2}\ mathbf {C} _ {2}\ mathbf {O} _ {4} (a) +2\ mathrm {Ag} ^ {2+} (a) +2\ mathrm {H} _ {2}\ mathrm {L} (л)\ правий лівий газон\\ 2\ текст {C}} _2 (g) + 2\ текст {Ag} ^+ (aq) + 2\ текст {H} _3\ текст {O} ^+ (aq)\) |
| Бр — | \(2\mathrm{Br}^{-} \rightleftharpoons \textbf{Br}_\textbf{2}+2 e^{-}\) | \ (\ textbf {H} _\ textbf {2}\ textbf {S} (a) +\ текст {Br} _ {2} (\ математика {aq}) +2\ mathrm {H} _ {2}\ mathrm {O} (\ mathrm {l})\ правий лівий гарпуни\ \ текст {S} (s) + 2\ текст {Br} ^- (aq) + 2\ текст {H} _3\ текст {O} ^+ (aq)\) |
| Се 3+ | \(\mathrm{Ce}^{3+} \rightleftharpoons \textbf{Ce}^\textbf{4+}+e^{-}\) |
\(\textbf{Fe}(\mathbf{C N})_\textbf{6}^\textbf{4–}(a q)+\text{ Ce}^{4+}(a q) \rightleftharpoons \\ \mathrm{Fe}(\mathrm{CN})_{6}^{3-}(a q)+\text{ Ce}^{3+}(a q)\) |
| Cl — | \(2\mathrm{Cl}^{-} \rightleftharpoons \textbf{Cl}_\textbf{2}+2 e^{-}\) | \(\textbf{Ti(I)}(a q)+\text{ Cl}_{2}(a q) \rightleftharpoons \mathrm{Ti}(\mathrm{III})(a q)+2 \mathrm{Cl}^{-}(a q)\) |
| Фе 3+ | \(\mathrm{Fe}^{3+} +e^{-} \rightleftharpoons \textbf{Fe}^\textbf{2+}\) | \ (\ mathbf {Cr} _\ textbf {2}\ mathbf {O} _\ textbf {7} ^\ mathbf {2-} (a) +6\ математика {Fe} ^ {2+} (a q) +14\ математика {H} _ {3}\ математика {O} ^ {+} (a q) правий лівий гарпуни\\ 2\ текст {Cr} ^ {3+} (aq) + 6\ текст {Fe} ^ {3+} (aq) + 21\ текст {H} _2\ text {O} (l)\) |
| Я — | \(3\mathrm{I}^{-} \rightleftharpoons \textbf{I}_\textbf{3}^\textbf{–}+2 e^{-}\) | \(2 \mathbf{S}_\mathbf{2} \mathbf{O}_\mathbf{3}^\mathbf{2-}(a q)+\mathrm{I}_{3}^{-}(a q) \rightleftharpoons \text{S}_{4} \mathrm{O}_{6}^{2-}(a q)+3 \mathrm{I}^{-}(a q)\) |
| Мн 2+ | \(\mathrm{Mn}^{2+} \rightleftharpoons \textbf{Mn}^\textbf{3+}+e^{-}\) | \(\textbf{As(III)}(a q)+2 \text{Mn}^{3+}(aq) \rightleftharpoons \text{As(V)}(a q)+2 \text{Mn}^{2+}(a q)\) |
|
Примітка: Електрохімічно генерований реагент і аналіт показані жирним шрифтом. |
||
Для аналіту, який нелегко окислювати або зменшити, ми можемо завершити кулометричне титрування шляхом з'єднання окислення або відновлення медіатора з кислотно-основою, осадженням або реакцією комплексоутворення, яка включає аналіт. Наприклад, якщо ми використовуємо H 2 O як медіатор, ми можемо генерувати H 3 O + на аноді
\[6 \mathrm{H}_{2} \mathrm{O}(l) \rightleftharpoons 4 \mathrm{H}_{3} \text{O}^{+}(a q)+\text{ O}_{2}(g)+4 e^{-} \nonumber\]
і генерувати OH — на катоді.
\[2 \mathrm{H}_{2} \mathrm{O}(l)+2 e^{-} \rightleftharpoons 2 \mathrm{OH}^{-}(a q)+\text{ H}_{2}(g) \nonumber\]
Якщо ми проводимо окислення або відновлення H 2 O за допомогою генераторної комірки на рис. Template:index, то ми можемо вибірково дозувати H 3 O + або OH — у розчин, який містить аналіт. Отримана реакція ідентична реакції при кислотно-лужному титруванні. Кулометричні кислотно-лужні титрування використовувались для аналізу сильних і слабких кислот і основ як у водних, так і в неводних матрицях. У таблиці Template:index узагальнено декілька прикладів кулометричних титрувань, які включають реакції кислотно-основи, комплексоутворення та осадження.
| тип реакції | посередник | електрохімічно генерований реагент і реакція | представник додаток |
|---|---|---|---|
| кислотно-лужний | Н 2 О | \(6 \mathrm{H}_{2} \mathrm{O} \rightleftharpoons 4 \textbf{H}_\mathbf{3} \textbf{O}^\mathbf{+}+\text{ O}_{2}+e^{-}\) | \(\textbf{OH}^\mathbf{-}(a q)+\text{ H}_{3} \mathrm{O}^{+}(a q) \rightleftharpoons 2 \mathrm{H}_{2} \mathrm{O}(l)\) |
| кислотно-лужний | Н 2 О | \(2 \mathrm{H}_{2} \mathrm{O}+2 e^{-}\rightleftharpoons 2 \textbf{OH}^\mathbf{-}+\text{ H}_{2}\) | \(\textbf{H}_\mathbf{3} \textbf{O}^\mathbf{+}(a q)+\text{ OH}^{-}(a q) \rightleftharpoons 2 \mathrm{H}_{2} \mathrm{O}(l)\) |
| комплексоутворення | ГнН 3 Y 2- (Y = ЕДТА) | \(\mathrm{HgNH}_{3} \mathrm{Y}^{2-}+\text{ NH}_{4}^{+} + 2 e^{-} \rightleftharpoons \\ \textbf{HY}^\mathbf{3-}+\text{ Hg}+2 \mathrm{NH}_{3}\) | \(\mathbf{Ca}^\mathbf{2+}(a q)+ \text{ HY}^{3-}(a q)+ \text{ H}_{2} \text{O}(l)\rightleftharpoons \\ \text{CaY}^{2-}(a q)+ \text{ H}_{3} \text{O}^{+}(a q)\) |
| комплексоутворення | Ag | \(\mathrm{Ag} \rightleftharpoons \textbf{ Ag}^\mathbf{+}+e^{-}\) | \(\mathbf{I}^\mathbf{-}(a q)+\text{ Ag}^{+}(a q) \rightleftharpoons \operatorname{Ag} \mathrm{I}(s)\) |
| опади | Hg | \(2 \mathrm{Hg} \rightleftharpoons \mathbf{H} \mathbf{g}_{2}^{2+}+2 e^{-}\) | \(2 \textbf{Cl}^\mathbf{-}(a q)+\text{ Hg}_{2}^{2+}(a q) \rightleftharpoons \text{ Hg}_{2} \mathrm{Cl}_{2}(s)\) |
| опади | \(\text{Fe(CN)}_6^{3-}\) | \(\mathrm{Fe}(\mathrm{CN})_{6}^{3-}+e^{-}\rightleftharpoons \textbf{ Fe(CN)}_\mathbf{6}^\mathbf{4-}\) | \(3 \mathbf{Zn}^\mathbf{2+}(a q)+ \text{K}^{+}(a q) +2 \text{Fe(CN)}_{6}^{4-}(a q) \rightleftharpoons \\ \text{K}_{2} \text{Zn}_{3}\left[\text{Fe(CN)}_{6}\right]_{2}(s) \) |
| Примітка: Електрохімічно генерований реагент і аналіт показані жирним шрифтом. | |||
У порівнянні зі звичайним титруванням, кулометричне титрування має дві важливі переваги. Перша перевага полягає в тому, що електрохімічна генерація титранту дозволяє нам використовувати нестійкий реагент. Хоча ми не можемо приготувати та зберігати розчин високореактивного реагенту, такого як Ag 2 + або Mn 3 +, ми можемо генерувати їх електрохімічним шляхом та використовувати їх в кулометричному титруванні. По-друге, оскільки відносно легко виміряти невелику кількість заряду, ми можемо використовувати кулометричне титрування для визначення аналіту, концентрація якого занадто мала для звичайного титрування.
Кількісні розрахунки
Абсолютна кількість аналіту в кулометричному аналізі визначається за законом Фарадея (Equation\ ref {11.1}) і сумарний заряд, заданий рівнянням\ ref {11.2} або рівнянням\ ref {11.3}. Наступний приклад показує розрахунки для типового кулометричного аналізу.
Для визначення чистоти зразка Na 2 S 2 O 3 зразок титрують кулометрично, використовуючи I — як медіатор і\(\text{I}_3^-\) як титрант. Пробу вагою 0,1342 г переносять в об'ємну колбу об'ємом 100 мл і розводять до об'єму дистильованою водою. На електрохімічну комірку переносять порцію 10,00 мл разом з 25 мл 1 М КІ, 75 мл фосфатного буфера pH 7,0 і декількома краплями розчину індикатора крохмалю. Електроліз при постійному струмі 36,45 мА вимагає 221,8 с для досягнення кінцевої точки показника крохмалю. Визначте чистоту зразка.
Рішення
Як показано в таблиці Template:index, кулометричне титрування\(\text{S}_2 \text{O}_3^{2-}\) with\(\text{I}_3^-\) дорівнює
\[2 \mathrm{S}_{2} \mathrm{O}_{3}^{2-}(a q)+\text{ I}_{3}^{-}(a q)\rightleftharpoons \text{ S}_{4} \mathrm{O}_{6}^{2-}(a q)+3 \mathrm{I}^{-}(a q) \nonumber\]
Для окислення\(\text{S}_2 \text{O}_3^{2-}\) до\(\text{S}_4 \text{O}_6^{2-}\) потрібно один електрон на\(\text{S}_2 \text{O}_3^{2-}\) (n = 1). Поєднання рівняння\ ref {11.1} та рівняння\ ref {11.2} та розв'язування молів і грам Na 2 S 2 O 3 дає
\[N_{A} =\frac{i t_{e}}{n F}=\frac{(0.03645 \text{ A})(221.8 \text{ s})}{\left(\frac{1 \text{ mol } e^{-}}{\text{mol Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3}}\right)\left(\frac{96487 \text{ C}}{\text{mol } e^{-}}\right)} =8.379 \times 10^{-5} \text{ mol Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3} \nonumber\]
Це кількість Na 2 S 2 O 3 в 10,00-мл порції 100-мл зразка; таким чином, в вихідному зразку є 0,1325 грам Na 2 S 2 O 3. Таким чином, чистота зразка є
\[\frac{0.1325 \text{ g} \text{ Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3}}{0.1342 \text{ g} \text { sample }} \times 100=98.73 \% \text{ w} / \text{w } \mathrm{Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3} \nonumber\]
Зауважимо, що для Equation\ ref {11.1} і Equation\ ref {11.2} не має значення, чи\(\text{S}_2 \text{O}_3^{2-}\) окислюється на робочому електроді або окислюється\(\text{I}_3^-\).
Для аналізу латунного сплаву зразок 0,442-г розчиняють в кислоті і розводять до об'єму в об'ємній колбі об'ємом 500 мл. Електроліз зразка 10,00 мл при —0,3 В проти SCE зменшує Cu 2 + до Cu, вимагаючи загального заряду 16,11 С. Регулювання потенціалу до —0,6 В проти SCE і завершення електролізу вимагає 0,442 С для зменшення Pb 2 + до Pb. Повідомити про %w/w Cu та Pb у сплаві.
- Відповідь
-
Для зменшення Cu 2 + до Cu потрібно два електрони на моль Cu (n = 2). Використовуючи Equation\ ref {11.1}, ми обчислюємо молі та грами Cu в аналізованій частині зразка.
\[N_{C u}=\frac{Q}{n F}=\frac{16.11 \text{ C}}{\frac{2 \text{ mol } e^{-}}{\mathrm{mol} \text{ Cu}} \times \frac{96487 \text{ C}}{\text{ mol } e^{-}}}=8.348 \times 10^{-5} \text{ mol Cu} \nonumber\]
\[8.348 \times 10^{-5} \text{ mol Cu} \times \frac{63.55 \text{ g Cu} }{\text{mol Cu}}=5.301 \times 10^{-3} \text{ g Cu} \nonumber\]
Це Cu з 10,00 мл частини зразка 500,0 мл; таким чином, %/w/w міді в вихідному зразку латуні є
\[\frac{5.301 \times 10^{-3} \text{ g Cu} \times \frac{500.0 \text{ mL}}{10.00 \text{ mL}}}{0.442 \text{ g sample} } \times 100=60.0 \% \text{ w/w Cu} \nonumber\]
Для свинцю ми дотримуємося того ж процесу; таким чином
\[N_{\mathrm{Pb}}=\frac{Q}{n F}=\frac{0.422 \text{ C}}{\frac{2 \text{ mol } e^-}{\text{mol Pb}} \times \frac{96487 \text{ C}}{\text{mol } e^{-}}}=2.19 \times 10^{-6} \text{ mol Pb} \nonumber\]
\[2.19 \times 10^{-6} \text{ mol Pb}\times \frac{207.2 \text{ g Pb} }{\text{mol Cu} }=4.53 \times 10^{-4} \text{ g Pb} \nonumber\]
\[\frac{4.53 \times 10^{-4} \text{ g Pb} \times \frac{500.0 \text{ mL}}{10.00 \text{ mL}}}{0.442 \text{ g sample}} \times 100=5.12 \% \text{ w/w Pb} \nonumber\]
Представницький метод 11.3.1: Визначення дихромату кулометричним окислювально-відновним титруванням
Найкращий спосіб оцінити теоретичні та практичні деталі, розглянуті в цьому розділі, - це уважно вивчити типовий аналітичний метод. Хоча кожен метод унікальний, наступний опис визначення\(\text{Cr}_2 \text{O}_7^{2-}\) дає повчальний приклад типової процедури. Опис тут базується на Бассетт, Дж.; Денні, Р.К.; Джеффрі, Г. Х.; Мендхем, Підручник кількісного неорганічного аналізу Дж. Фогеля, Лонгман: Лондон, 1978, стор. 559—560.
Опис методу
Концентрацію\(\text{Cr}_2 \text{O}_7^{2-}\) в зразку визначають кулометричним окислювально-відновним титруванням з використанням Fe 3 + як медіатора і електрогенірованого Fe 3 + як титранта. Кінцева точка титрування визначається потенціометрично.
Порядок дій
Електрохімічна комірка складається з робочого електрода Pt і зустрічного електрода Pt, розміщених в окремих осередках, з'єднаних пористим скляним диском. Заповніть комірку зустрічного електрода 0,2 M Na 2 SO 4, зберігаючи рівень вище рівня розчину в комірці робочого електрода. Підключіть платиновий електрод і вольфрамовий електрод до потенціометра, щоб ви могли виміряти потенціал робочого електрода під час аналізу. Приготуйте розчин медіатора приблизно 0,3 М NH 4 Fe (SO 4) 2. Додайте 5,00 мл зразка, 2 мл 9 M H 2 SO 4 та 10-25 мл розчину медіатора до осередку робочого електрода та додайте дистильовану воду, якщо потрібно, щоб покрити електроди. Пузир чистий N 2 через розчин протягом 15 хв для видалення будь-якого O 2, який присутній. Підтримуйте потік N 2 під час електролізу, вимикаючи його на мить при вимірюванні потенціалу. Перемішайте розчин, використовуючи магнітну смужку для перемішування. Налаштуйте струм до 15-50 мА і починайте титрування. Періодично припиняйте титрування і вимірюйте потенціал. Побудувати криву титрування потенціалу в порівнянні з часом і визначити час, необхідний для досягнення точки еквівалентності.
Запитання
1. Платиновий робочий електрод - катод або анод?
Зниження Fe 3 + до Fe 2 + відбувається на робочому електроді, роблячи його катодом в цій електрохімічній осередку.
2. Чому необхідно видаляти розчинений кисень шляхом барботування N 2 через розчин?
Будь-який розчинений O 2 окислює Fe 2 + назад до Fe 3 +, як показано наступною реакцією.
\[4\text{Fe}^{2+}(aq) + \text{ O}_2 + \text{ 4H}_3\text{O}^+(aq) \rightleftharpoons 4\text{Fe}^{3+}(aq) + 6\text{H}_2\text{O}(l) \nonumber\]
Для підтримки ефективності струму всі Fe 2 + повинні реагувати з\(\text{Cr}_2 \text{O}_7^{2-}\). Реакція Fe 2 + з O 2 означає, що потрібно більше медіатора Fe 3 +, збільшуючи час досягнення кінцевої точки титрування. В результаті повідомляємо про наявність занадто багато\(\text{Cr}_2 \text{O}_7^{2-}\).
3. Який вплив на аналіз, якщо NH 4 Fe (SO 4) 2 забруднений слідовими кількостями Fe 2 +? Як можна компенсувати це джерело Fe 2 +?
Існує два джерела Fe 2 +: що генерується посередником і присутній як домішка. Оскільки загальна кількість Fe 2 +, з яким реагує,\(\text{Cr}_2 \text{O}_7^{2-}\) залишається незмінним, від медіатора потрібно менше Fe 2 +. Це зменшує час, необхідний для досягнення кінцевої точки титрування. Оскільки очевидна ефективність струму перевищує 100%, повідомлена\(\text{Cr}_2 \text{O}_7^{2-}\) концентрація занадто мала. Ми можемо видалити слідову кількість Fe 2 + з розчину медіатора шляхом додавання H 2 O 2 і нагрівання при 50-70 o С до припинення еволюції O 2, перетворюючи Fe 2 + в Fe 3 +. Крім того, ми можемо виконати порожнє титрування, щоб виправити будь-які домішки Fe 2 + в медіаторі.
4. Чому рівень розчину в комірці зустрічного електрода підтримується вище рівня розчину в комірці робочого електрода?
Це запобігає потраплянню розчину, який містить аналіт, в комірку зустрічного електрода. Окислення Н 2 О на зустрічному електроді виробляє О 2, який може вступати в реакцію з Fe 2 +, що утворюється на робочому електроді або Cr 3 +, що виникає в результаті реакції Fe 2 + і\(\text{Cr}_2 \text{O}_7^{2-}\). У будь-якому випадку результат є позитивною детермінантною помилкою.
Характеристика додатків
Одним з корисних застосувань кулометрії є визначення кількості електронів, що беруть участь в окисно-відновній реакції. Для визначення ми завершуємо кулометричний аналіз з контрольованим потенціалом, використовуючи відому кількість чистої сполуки. Сумарний заряд в кінці електролізу використовується для визначення значення n за законом Фарадея (Equation\ ref {11.1}).
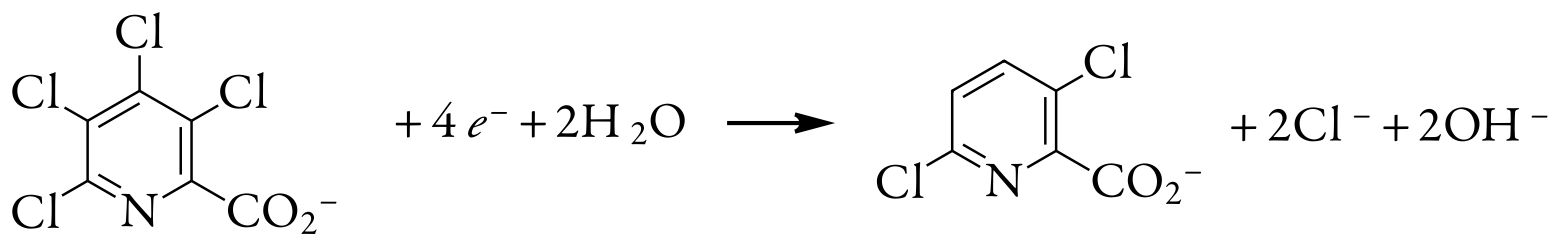
0,3619-г зразка тетрахлоропіколінової кислоти, C 6 HNO 2 Cl 4, розчиняють у дистильованій воді, переносять у об'ємну колбу об'ємом об'ємом 1000 мл і розводять до об'єму. Для вичерпного електролізу з контрольованим потенціалом 10,00-мл порції цього розчину на губчастому срібному катоді потрібно 5,374 С заряду. Яке значення n для цієї реакції відновлення?
Рішення
10,00-мл частина зразка містить 3,619 мг, або\(1.39 \times 10^{-5}\) моль тетрахлоропіколінової кислоти. Розв'язування рівняння\ ref {11.1} для n і внесення відповідних замін дає
\[n=\frac{Q}{F N_{A}}=\frac{5.374 \text{ C}}{\left(96478 \text{ C/mol } e^{-}\right)\left(1.39 \times 10^{-5} \text{ mol } \mathrm{C}_{6} \mathrm{HNO}_{2} \mathrm{Cl}_{4}\right)} = 4.01 \text{ mol e}^-/\text{mol } \mathrm{C}_{6} \mathrm{HNO}_{2} \mathrm{Cl}_{4} \nonumber\]
Таким чином, для відновлення молекули тетрахлоропіколінової кислоти потрібно чотири електрони. Загальна реакція, в результаті якої відбувається селективне утворення 3,6-дихлоропіколінової кислоти, становить
Оцінка
Масштаб операції
Кулометричний метод аналізу може аналізувати невелику абсолютну кількість аналіта. У кулометрії з контрольованим струмом, наприклад, молі аналіту, споживаного під час вичерпного електролізу, задаються рівнянням\ ref {11.11}. Електроліз з використанням постійного струму 100 мкА протягом 100 с, наприклад, споживає тільки\(1 \times 10^{-7}\) моль аналіту, якщо n = 1. Для аналіта з молекулярною масою 100\(1 \times 10^{-7}\) г/моль аналіту відповідає всього 10 мкг. Концентрація аналіту в електрохімічній комірці, однак, повинна бути достатньою для точного визначення кінцевої точки. При використанні візуальної кінцевої точки найменша концентрація аналіту, яку можна визначити кулометричним титруванням, становить приблизно 10 —4 М. Як і у випадку зі звичайним титруванням, кулометричне титрування з використанням візуальної кінцевої точки обмежується основними і незначними аналітами. Кулометричне титрування до заданої потенціометричної кінцевої точки можливо, навіть якщо концентрація аналіта настільки мала, як 10 —7 М, розширюючи аналіз для відстеження аналітів [Curran, DJ «Кулометрія постійного струму», в Кіссінджер, П.Т.; Heineman, W.R., ред., Лабораторні методи в Електроаналітична хімія, Марсель Деккер Інк.: Нью-Йорк, 1984, с. 539—568].
Точність
У кулометрії з контрольованим струмом точність визначається точністю, з якою ми можемо виміряти струм і час, і точністю, з якою ми можемо ідентифікувати кінцеву точку. Максимальні похибки вимірювань по струму і часу складають близько ± 0,01% і ± 0,1% відповідно. Максимальна похибка кінцевої точки для кулометричного титрування принаймні така ж хороша, як і для звичайного титрування, і часто краще при використанні невеликих кількостей реагентів. Разом ці похибки вимірювань дозволяють припустити, що точність 0,1% - 0,3% можлива. Отже, обмежуючим фактором у багатьох аналізах є поточна ефективність. Поточний ККД понад 99,5% є досить рутинним, і він часто перевищує 99,9%.
У кулометрії з керованим потенціалом точність визначається коефіцієнтом корисної дії струму і визначенням заряду. Якщо зразок не містить перешкод, які легше окислювати або зменшувати, ніж аналіт, ефективність струму більше 99,9% є звичайною. Коли присутній інтерферент, його часто можна усунути шляхом застосування потенціалу, коли вичерпний електроліз перешкод можливий без одночасного електролізу аналіту. Після того, як інтерферент видаляється, потенціал перемикається на рівень, де можливий електроліз аналіту. Граничним фактором точності багатьох кулометричних методів аналізу з керованим потенціалом є визначення заряду. З електронними інтеграторами сумарний заряд визначається з точністю краще 0,5%.
Якщо ми не можемо отримати прийнятну ефективність струму, електрогравіметричний аналіз можливий, якщо аналіт - і тільки аналіт - утворює твердий наліт на робочому електроді. При цьому робочий електрод зважується перед початком електролізу і переважується по завершенні електролізу. Різниця в вазі електрода дає масу аналіта.
Точність
Точність визначається невизначеністю вимірювання струму, часу та кінцевої точки в кулометрії з керованим струмом або зарядом в кулометрії з керованим потенціалом. Точність ± 0,1— 0,3% отримується регулярно при кулометричному титруванні, а точність ± 0,5% характерна для кулометрії з керованим потенціалом.
Чутливість
Для кулометричного методу аналізу калібрувальна чутливість еквівалентна nF у Equation\ ref {11.1}. Взагалі, кулометричний метод є більш чутливим, якщо окислення або відновлення аналіта передбачає більшу величину n.
Вибірковість
Вибірковість в кулометрії з керованим потенціалом та керованим струмом покращується шляхом регулювання умов розчину та вибору потенціалу електролізу. У кулометрії з керованим потенціалом потенціал фіксується потенціостатом, а в кулометрії з керованим струмом потенціал визначається окисно-відновною реакцією з медіатором. У будь-якому випадку можливість контролю потенціалу електролізу дає певну міру селективності. Регулюючи рН або додаючи комплексообразователь, можна зрушити потенціал, при якому аналіт або інтерферент піддається окисленню або відновленню. Наприклад, потенціал зниження стандартного стану для Zn 2 + становить —0,762 В проти SHE. Якщо додати розчин NH 3, утворюючи\(\text{Zn(NH}_3\text{)}_4^{2+}\), потенціал стандартного стану зміщується до —1,04 В. це забезпечує додатковий засіб для контролю селективності, коли аналіт і інтерферент піддаються електролізу при аналогічних потенціалах.
Час, вартість та обладнання
Кулометрія з контрольованим потенціалом є відносно трудомістким аналізом, при цьому типовий аналіз вимагає 30-60 хв. Кулометричні титрування, з іншого боку, вимагають всього декількох хвилин, і їх легко адаптувати до автоматизованого аналізу. Комерційні прилади як для кулометрії з керованим потенціалом, так і для контрольованого струму доступні, і є відносно недорогими. Низькі потенціостати та джерела постійного струму доступні приблизно за 1000 доларів.