11.5: Проблеми
- Page ID
- 24951

1. Визначте анод і катод для наступних електрохімічних елементів та визначте реакцію окислення або відновлення на кожному електроді.
(а) Пт | FeCl 2 (ак, 0,015), FeCl 3 (ак, 0,045) || АгО 3 (ак, 0,1) | Ag
(б) Аг | АгБр (и), NaBr (ак, 1,0) || ЦСл 2 (ак, 0,05) | Кд
(с) Пб | ПбСО 4 (и), Н 2 СО 4 (ак, 1.5) |Н 2 СО 4 (ак, 2.0), ПбСО 4 (и) | ПБО 2
2. Обчисліть потенціал для кожного електрохімічного елемента в задачі 1. Значення в дужках - це діяльність асоційованого виду.
3. Обчисліть активність КІ, х, в наступному електрохімічному осередку, якщо потенціал дорівнює +0,294 В.
Аг | АГкл (и), NaCl (ак, 0,1) || К (ак, х), I 2 (и) | Пт
4. Яка реакція заважає нам використовувати Zn як електрод першого роду в кислому розчині? Які ще метали ви очікуєте вести себе так само, як Zn при зануренні в кислий розчин?
5. Creager і його колеги розробили саліцилатний іонселективний електрод з використанням ПВХ-мембрани, просоченої саліцилатом тетраалкіламмонію [Creager, S.E.; Lawrence, K.D.; Tibbets, C. RJ Chem. Едук. 1995, 72, 274—276]. Для визначення коефіцієнта селективності іоноселективного електрода для бензоату підготували набір стандартів калібрування саліцилатів, в яких концентрація бензоату трималася постійною на рівні 0,10 М. Використовуючи наступні дані, визначають значення коефіцієнта селективності.
| [саліцилат] (М) | потенціал (мВ) |
|---|---|
| 1.0 | 20.2 |
| \(1.0 \times 10^{-1}\) | 73.5 |
| \(1.0 \times 10^{-2}\) | 126 |
| \(1.0 \times 10^{-3}\) | 168 |
| \(1.0 \times 10^{-4}\) | 182 |
| \(1.0 \times 10^{-5}\) | 182 |
| \(1.0 \times 10^{-6}\) | 177 |
Яка максимально допустима концентрація бензоату, якщо ви плануєте використовувати цей іоноселективний електрод для аналізу зразка, який містить всього 10 -5 М саліцилату з точністю краще 1%?
6. Ватанабе та колеги описали новий мембранний електрод для визначення кокаїну, слабкого базового алкалоїду з р К а 8,64 [Ватанабе, К.; Oda, K.; Oda, H.; Furuno, K.; Gomita, Y.; Katsu, T. Чим. Акт 1995, 316, 371—375]. Відповідь електрода на фіксовану концентрацію кокаїну не залежить від рН в діапазоні 1-8, але різко знижується вище рН 8. Запропонуйте пояснення цієї залежності рН.
7. На малюнку 11.2.14 показана принципова схема ферментного електрода, який реагує на сечовину, використовуючи газочутливий електрод NH 3 для вимірювання кількості аміаку, що виділяється після реакції ферменту з сечовиною. У свою чергу, електрод NH 3 використовує pH-електрод для контролю зміни рН за рахунок аміаку. Відгук електрода сечовини задається рівнянням 11.2.12. Починаючи з рівняння 11.2.19, яке дає потенціал pH-електрода, показують, що рівняння 11.2.12 для електрода сечовини правильне.
8. Поясніть, чому реакція електрода сечовини на основі NH 3 (рис. 11.2.14 та рівняння 11.2.12) відрізняється від реакції електрода сечовини, в якому фермент покритий скляною мембраною pH-електрода (рис. 11.2.15 та рівняння 11.2 .13).
9. Потенціометричний електрод для HCN використовує газопроникну мембрану, буферний внутрішній розчин 0,01 М KAg (CN) 2 і електрод Ag 2 S ISE, який занурений у внутрішній розчин. Розглянемо реакції рівноваги, що відбуваються всередині внутрішнього розчину, і виведіть рівняння, яке пов'язує потенціал електрода з концентрацією HCN у зразку. Щоб перевірити свою роботу, шукайте в режимі он-лайн патент США 3859191 та зверніться до малюнка 2.
10. Міффлін та асоційовані описали мембранний електрод для кількісного аналізу пеніциліну, в якому фермент пеніциліназа іммобілізується в поліакриламідному гелі, нанесеному на скляну мембрану pH-електрода [Mifflin, T. E.; Andriano, K.M.; Robbins, W.B. J. Chem. Едук. 1984, 61, 638—639]. Наступні дані були зібрані з використанням набору норм пеніциліну.
| [пеніцилін] (М) | потенціал (мВ) |
|---|---|
| \(1.0 \times 10^{-2}\) | 220 |
| \(2.0 \times 10^{-3}\) | 204 |
| \(1.0 \times 10^{-3}\) | 190 |
| \(2.0 \times 10^{-4}\) | 153 |
| \(1.0 \times 10^{-4}\) | 135 |
| \(1.0 \times 10^{-5}\) | 96 |
| \(1.0 \times 10^{-6}\) | 80 |
(а) За яким діапазоном концентрацій існує лінійна реакція?
(b) Яке рівняння калібрувальної кривої для цього діапазону концентрацій?
(c) Яка концентрація пеніциліну у зразку, який дає потенціал 142 мВ?
11. Іоноселективний електрод може бути поміщений в проточну комірку, в яку ми впорскуємо зразки або стандарти. Коли аналіт проходить через клітину, замість стійкого потенціалу реєструється потенційний сплеск. Концентрація К+ в сироватці крові була визначена таким чином, використовуючи стандарти, підготовлені в матриці 0,014 M NaCl [Meyerhoff, M.E.; Kovach, P.MJ Chem. Едук. 1983, 9, 766—768].
| [К+] (мМ) | E (арб. одиниці) | [К+] (мМ) | E (арб. одиниці) |
|---|---|---|---|
| 0,10 | 25.5 | 0,60 | 58.7 |
| 0,20 | 37.2 | 0,80 | 64.0 |
| 0,40 | 50.8 | 1.00 | 66.8 |
1,00-мл зразка сироватки розводять до об'єму в об'ємній колбі об'ємом 10 мл і аналізують, даючи потенціал 51,1 (довільні одиниці). Повідомити про концентрацію К + в зразку сироватки крові.
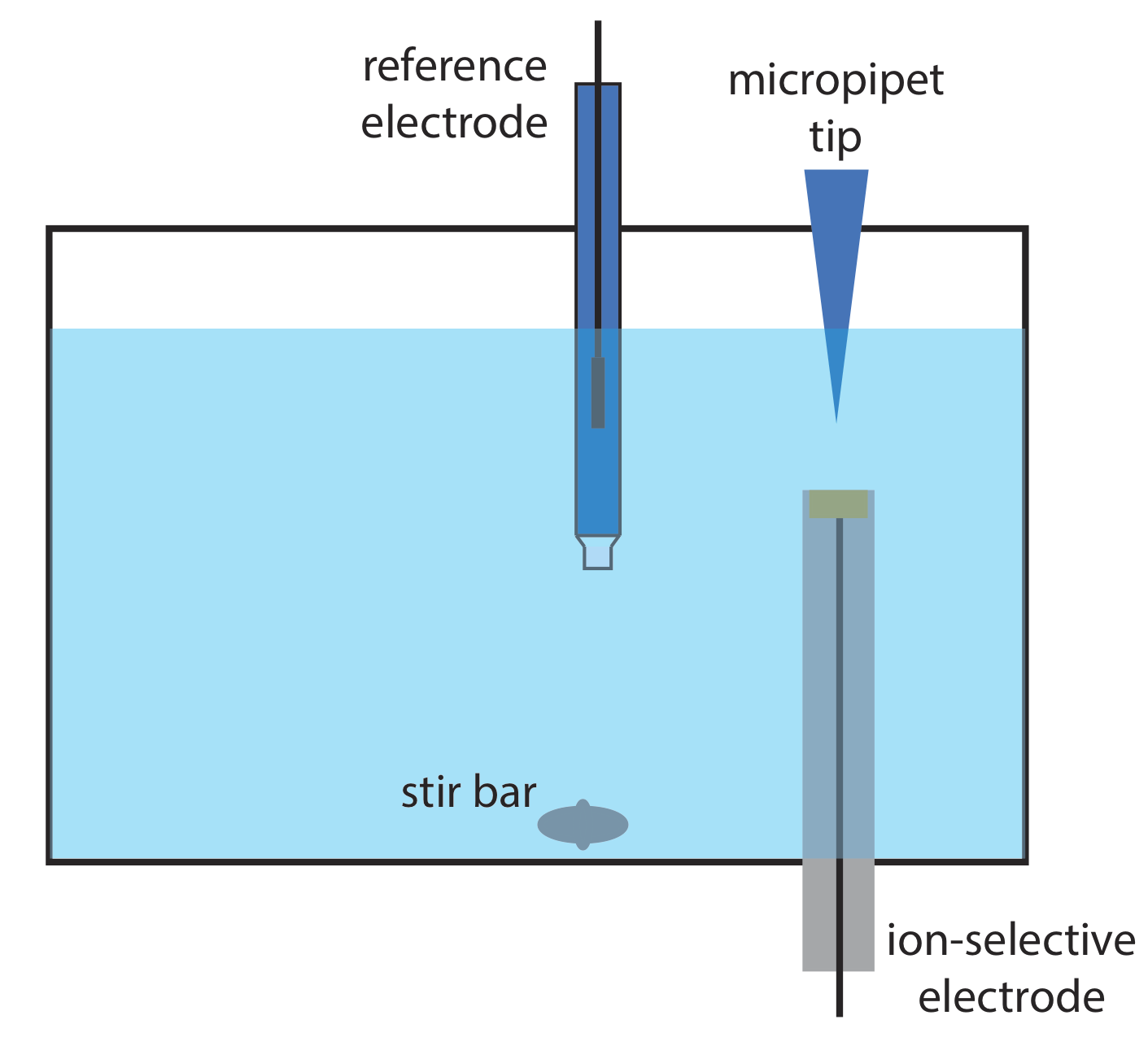
12. Ван і Таха описали цікаве застосування потенціометрії, яку вони називають пакетною ін'єкцією [Ван, Дж.; Таха, З. анал. Чим. Акт 1991, 252, 215—221]. Як показано на малюнку нижче, іоноселективний електрод поміщають в перевернутому положенні в резервуарі великого об'єму, а фіксований обсяг зразка або стандартного розчину вводять до поверхні електрода за допомогою мікропіпета. Відповідь електрода - це сплеск потенціалу, пропорційний концентрації аналіта. Наступні дані були зібрані за допомогою електрода pH і набору стандартів рН.
| рН | потенціал (мВ) |
|---|---|
| 2.0 | +300 |
| 3.0 | +240 |
| 4.0 | +168 |
| 5.0 | +81 |
| 6.0 | +35 |
| 8.0 | —92 |
| 9.0 | —168 |
| 10.0 | —235 |
| 11.0 | —279 |
Визначають рН наступних зразків з урахуванням зафіксованих пікових потенціалів: томатний сік, 167 мВ; водопровідна вода, —27 мВ; кава - 122 мВ.
13. Концентрація\(\text{NO}_3^-\) в пробі води визначається одноточковим стандартним додаванням за допомогою\(\text{NO}_3^-\) іоноселективного електрода. Пробу об'ємом 25,00 мл поміщають у склянку і вимірюють потенціал 0,102 В. Додають 1,00-мл аліквоту 200,0-мг/л стандартного\(\text{NO}_3^-\) розчину, після чого потенціал становить 0,089 В. повідомити мг\(\text{NO}_3^-\) /л у пробі води.
14. У 1977 році, коли я був студентом коледжу Нокс, ми з моїм партнером лабораторії завершили експеримент з визначення концентрації фтору у водопровідній воді та кількості фтору в зубній пасті. Дані в цій проблемі взяті з мого лабораторного ноутбука.
(а) Для аналізу водопровідної води ми взяли три 25,0-мл проби і додали 25,0 мл TISAB до кожного. Потенціал кожного розчину вимірювали за допомогою F — ISE та опорного електрода SCE. Далі ми зробили п'ять додавань по 1,00-мл стандартного розчину по 100,0 проміле F — до кожного зразка, і вимірювали потенціал після кожного додавання, тричі фіксуючи потенціал.
| мл стандартного доданого | потенціал (мВ), реплікація 1 | потенціал (мВ), реплікація 2 | потенціал (мВ), реплікація 3 |
|---|---|---|---|
| 0.00 | —79 | —82 | —83 |
| 1.00 | -119 | -119 | -18 |
| 2.00 | -133 | -133 | -133 |
| 3.00 | —142 | —142 | —142 |
| 4.00 | —149 | —148 | —148 |
| 5.00 | —154 | —153 | —153 |
Повідомити про частинки-мільйон F — у водопровідній воді.
(б) Для аналізу зубної пасти ми відміряли 0,3619 г у об'ємну колбу об'ємом 100 мл, додали 50,0 мл TISAB і розвели до об'єму дистильованою водою. Після того, як ми переконалися, що зразок був ретельно перемішаний, ми перенесли три порції по 20,0-мл в окремі склянки і виміряли потенціал кожної з них за допомогою F - ISE і опорного електрода SCE. Далі ми зробили п'ять додавань по 1,00-мл стандартного розчину по 100,0 проміле F — до кожного зразка, і вимірювали потенціал після кожного додавання, тричі фіксуючи потенціал.
| мл стандартного доданого | потенціал (мВ), реплікація 1 | потенціал (мВ), реплікація 2 | потенціал (мВ), реплікація 3 |
| 0.00 | -55 | —54 | -55 |
| 1.00 | —82 | —82 | —83 |
| 2.00 | —94 | —94 | —94 |
| 3.00 | —102 | —103 | —102 |
| 4.00 | —108 | —108 | —109 |
| 5.00 | -12 | -12 | -13 |
Повідомити про частинки-мільйон F — в зубній пасті.
15. Ви несете відповідальність за визначення кількості КІ в йодованій солі і вирішуєте використовувати I — іоноселективний електрод. Опишіть, як би ви виконували цей аналіз, використовуючи зовнішні стандарти, і як ви б сформували цей аналіз за допомогою методу стандартних доповнень.
16. Поясніть, чому кожне з наступних дій зменшує час аналізу в кулометрії з контрольованим потенціалом: більша площа поверхні для робочого електрода; менший об'єм розчину; і більш швидка швидкість перемішування.
17. Чистоту зразка пікринової кислоти, С 6 Н 3 Н 3 О 7, визначають за допомогою кулометрії з контрольованим потенціалом, перетворюючи пікринову кислоту в триамінофенол, C 6 H 9 N 3 O.
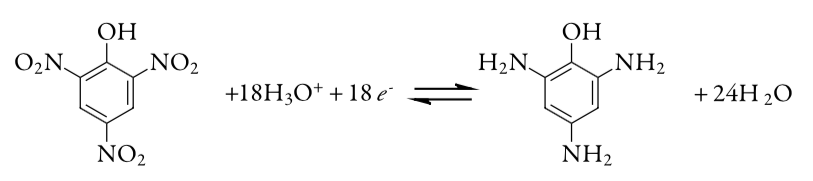
0,2917-г проби пікринової кислоти поміщають в об'ємну колбу об'ємом 1000 мл і розводять до об'єму. 10,00-мл порцію цього розчину переносять в кулометричну комірку і додають достатню кількість води, щоб катод Pt був занурений. Для вичерпного електролізу зразка потрібно 21,67 С заряду. Повідомити про чистоту пікринової кислоти.
18. Концентрацію Н 2 S в дренажі з покинутої шахти визначають кулометричним титруванням з використанням КІ як медіатора і\(\text{I}_3^-\) як титранта.
\[\text{H}_{2}\text{S}(a q)+\ \mathrm{I}_{3}^{-}(a q)+2 \mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons2 \mathrm{H}_{3} \mathrm{O}^{+}(a q)+3 \mathrm{I}^{-}(a q)+\mathrm{S}(s) \nonumber\]
У кулометричну осередок поміщається 50,00-мл проба води разом з надлишком КІ і невеликою кількістю крохмалю в якості показника. Електроліз проводять при постійному струмі 84,6 мА, що вимагає 386 с для досягнення кінцевої точки крохмалю. Повідомляють про концентрацію Н 2 S в зразку в мкг/мл.
19. Одним із способів визначення заданої маси H 3 SO 3 є кулометричне титрування, що використовується\(\text{I}_3^-\) як титрант. Відповідні реакції та потенціали стандартного стану узагальнені тут
\[\begin{aligned} \mathrm{H}_{3} \mathrm{AsO}_{4}(a q)+2 \mathrm{H}^{+}(a q)+2 \mathrm{e}^{-} &\rightleftharpoons \ \mathrm{H}_{3} \mathrm{AsO}_{3}(a q)+\ \mathrm{H}_{2} \mathrm{O}(l) \\ \mathrm{I}_{3}^{-}(a q)+2 \mathrm{e}^{-} &\rightleftharpoons 3 \mathrm{I}^{-}(a q) \end{aligned} \nonumber\]
при стандартному стані відновних потенціалів відповідно +0,559 В і +0,536 В. пояснити, чому кулометричне титрування проводять в нейтральному розчині (pH ≈ 7) замість в сильнокислому розчині (рН < 0).
20. Виробництво адипонітрилу, NC (CH 2) 4 CN, з акрилонітрилу, CH 2 = CHCN, є важливим промисловим процесом. 0,594-г зразок акрилонітрилу поміщають в об'ємну колбу об'ємом 1 л і розводять до об'єму. Для вичерпного електролізу з контрольованим потенціалом 1,00-мл порції розведеного акрилонітрилу потрібно 1,080 С заряду. Яке значення n для відновлення акрилонітрилу до адипонітрилу?
21. Гідродинамічна вольтаммограма лінійно-потенційного сканування для суміші Fe 2 + і Fe 3 + показана на малюнку нижче, де i l, a і i l, c - анодний і катодний граничні течії.
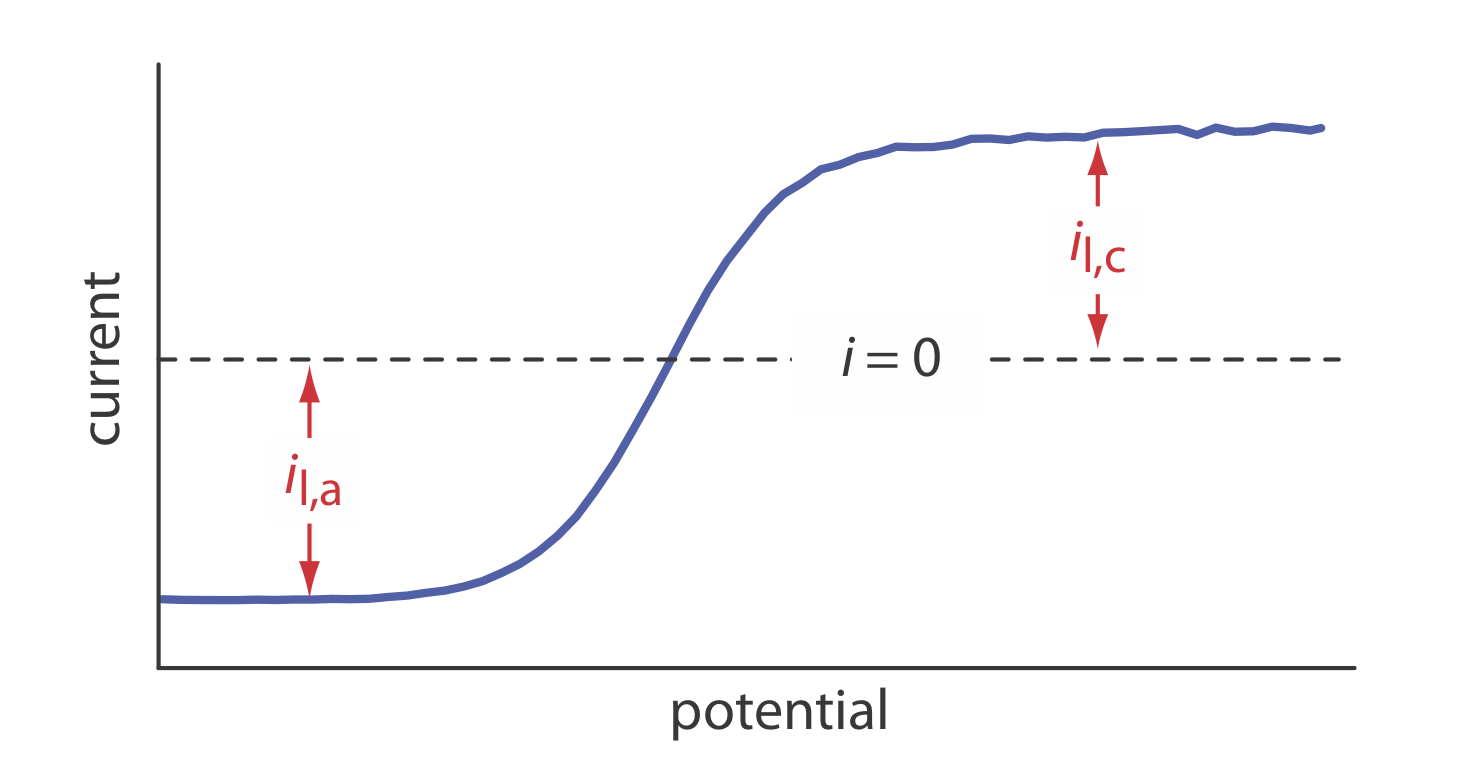
(а) Показати, що потенціал надається
\[E = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} - 0.05916 \log \frac {K_{\text{Fe}^{3+}}} {K_{\text{Fe}^{2+}}} - 0.05916 \log \frac {i - i_{l,a}}{i_{l,c} - i} \nonumber\]
(b) Який потенціал, коли i = 0 для розчину, який становить 0.100 мМ Fe 3 + і 0,050 мМ Fe 2 +?
22. Кількість сірки в ароматичних мономерах визначається за допомогою диференціальної імпульсної полярографії. Стандартні розчини готують для аналізу шляхом розчинення 1.000 мл очищеного мономера в 25,00 мл електролітичного розчинника, додавання відомої кількості сірки, деаерації, вимірювання пікового струму. Для набору стандартів калібрування отримані наступні результати.
| додано мкг S | піковий струм (мкА) |
|---|---|
| 0 | 0,14 |
| 28 | 0,70 |
| 56 | 1.23 |
| 112 | 2.41 |
| 168 | 3.42 |
Аналіз зразка об'ємом 1.000-мл, обробленого аналогічно стандартам, дає піковий струм 1,77 мкА. Повідомте про мг С/мл у зразку.
23. Чистоту зразка K 3 Fe (CN) 6 визначають за допомогою лінійно-потенційної скануючої гідродинамічної вольтамметрії на склоподібному вуглецевому електроді. Для набору зовнішніх стандартів калібрування отримані наступні дані.
| [K 3 Fe (CN) 6] (мМ) | граничний струм (мкА) |
|---|---|
| 2.0 | 127 |
| 4.0 | 252 |
| 6.0 | 376 |
| 8.0 | 500 |
| 10.0 | 624 |
Зразок нечистої К 3 Fe (CN) 6 готують до аналізу шляхом розведення 0,246-г порції до об'єму в об'ємній колбі об'ємом 100 мл. Граничний струм для зразка дорівнює 444 мкА. Повідомити про чистоту цього зразка K 3 Fe (CN) 6.
24. Одним із методів визначення того, чи людина нещодавно вистрілив пістолет, є пошук слідів сурми в залишках, зібраних з рук людини. Для цього аналізу ідеально підходить анодна зачистка вольтамметрія на ртутному плівковому електроді. У типовому аналізі зразок забирають у підозрюваного за допомогою ватного тампона, змоченого 5% v/v HNO 3. Після повернення в лабораторію тампон поміщають у флакон, який містить 5,0 мл 4 М HCl, що становить 0,02 М в сульфаті гідразину. Після замочування тампона 4,0-мл порцію розчину переносять в електрохімічну осередок разом зі 100 мкл 0,01 М HGCl 2. Після осадження тонкої плівки ртуті і сурми крок зачистки дає піковий струм 0,38 мкА. Після додавання стандартного додавання 100 мкл\(5.00 \times 10^2\) ppb Sb піковий струм збільшується до 1,14 мкА. Скільки нанограм Сб було зібрано з руки підозрюваного?
25. Цинк використовується як внутрішній стандарт при аналізі талію методом диференціальної імпульсної полярографії. Стандартний розчин\(5.00 \times 10^{-5}\) M Zn 2 + і\(2.50 \times 10^{-5}\) M Tl + має пікові струми 5,71 мкА і 3,19 мкА відповідно. 8,713-г зразок безцинкового сплаву розчиняють в кислоті, переносять в об'ємну колбу об'ємом 500 мл і розводять до об'єму. 25,0-мл порцію цього розчину змішують з 25,0 мл\(5.00 \times 10^{-4}\) M Zn 2 +. Аналіз цього розчину дає пікові струми 12,3 мкА і 20,2 мкА для Zn 2 + і Tl + відповідно. Повідомити про %w/w Tl у сплаві.
26. Диференціальна імпульсна вольтамметрія на вуглецевому робочому електроді використовується для визначення концентрацій аскорбінової кислоти та кофеїну в лікарських препаратах [Lau, O.; Luk, S.; Cheung, Y. Analyst 1989, 114, 1047—1051]. При типовому аналізі таблетку 0,9183-г подрібнюють і подрібнюють в дрібний порошок. 0,5630-г проби цього порошку переносять в об'ємну колбу об'ємом 100 мл, вносять в розчин і розводять до об'єму. Потім порцію цього розчину 0,500 мл переносять на вольтамметричну комірку, яка містить 20,00 мл відповідного підтримуючого електроліту. Отримана вольтаммограма дає пікові струми 1,40 мкА і 3,88 мкА для аскорбінової кислоти і для кофеїну відповідно. Потім додають 0,500-мл аліквоту стандартного розчину, який містить 250,0 ppm аскорбінової кислоти і 200,0 ppm кофеїну. Вольтаммограма цього розчину дає пікові струми 2,80 мкА і 8,02 мкА для аскорбінової кислоти і кофеїну відповідно. Повідомте міліграми аскорбінової кислоти і міліграми кофеїну в таблетці.
27. Ратана-ohpas та колеги описали метод аналізу зачистки для визначення олова в консервованих фруктових соках [Ратана-ohpas, R.; Kanatharana, P.; Ratana-ohpas, W.; Kongsawasdi, W. Anal. Чим. Акт 1996, 333, 115—118]. Були проаналізовані стандарти 50,0 ppb Sn 4 +, 100.0 ppb Sn 4 + та 150.0 ppb Sn 4 +, що дають пікові струми (довільні одиниці) відповідно 83,0, 171,6 та 260,2. 2.00-мл зразка соку лічі змішують з 20,00 мл 1:1 HCl/HnO 3. До 10 мл 6 М HCl додають порцію 0,500-мл цієї суміші і доводять обсяг до 30.00 мл. Аналіз цього розведеного зразка дав сигнал 128,2 (довільні одиниці). Повідомте про частини на мільйон Sn 4 + у вихідному зразку соку лічі.
28. Ситтампалам і Вілсон описали підготовку та використання амперометричного датчика для глюкози [Sittampalam, G; Wilson, G.SJ. Chem. Едук. 1982, 59, 70—73]. Датчик калібрується шляхом вимірювання сталого струму при зануренні його в стандартні розчини глюкози. Типовий набір даних калібрування наведено тут.
| [глюкоза] (мг/100 мл) | струм (арб. одиниць) |
|---|---|
| 2.0 | 17.2 |
| 4.0 | 32.9 |
| 6.0 | 52.1 |
| 8.0 | 68.0 |
| 10.0 | 85.8 |
Пробу 2,00 мл розводять до 10 мл в об'ємній колбі і вимірюють сталий струм 23,6 (довільні одиниці). Яка концентрація глюкози в пробі в мг/100 мл?
29. Диференціальна імпульсна полярографія використовується для визначення концентрацій свинцю, талію, індію в суміші. Оскільки піки для свинцю і талію, а для талію і індію перекриваються, необхідний одночасний аналіз. Пікові струми (в довільних одиницях) при —0,385 В, —0,455 В і —0,557 В вимірюють для єдиного стандартного рішення, а для вибірки, даючи результати, наведені в наступній таблиці. Повідомте про мг/мл Pb 2 +, Tl + та In 3 + у зразку.
| аналіт | [стандарт] (мкг/мл) | піковий струм при —0,385 В | піковий струм при —0,455 В | піковий струм при —0,557 В |
|---|---|---|---|---|
| Пб 2 + | 1.0 | 26.1 | 2.9 | 0 |
| Тл + | 2.0 | 7.8 | 23.5 | 3.2 |
| У 3+ | 0.4 | 0 | 0 | 22,9 |
| зразок | 60.6 | 28.8 | 54.1 | |
30. Абас та колеги розробили амперометричний біосенсор\(\text{NH}_4^+\), який використовує фермент глутаматдегідрогеназу для каталізації наступної реакції
\[2 \text { - oxyglutarate }(a q)+ \ \mathrm{NH}_{4}^{+}(a q)+\mathrm{NADH}(a q)\rightleftharpoons\text { glutamate }(a q)+\ \mathrm{NAD}^{+}(a q)+\ \mathrm{H}_{2} \mathrm{O}(l) \nonumber\]
де NADH є відновленою формою нікотинаміду аденіндинуклеотиду [Абасс, А.К.; Харт, Дж. П.; Коуелл, Д. С.; Чапелл, А. анальний. Чим. Акт 1988, 373, 1—8]. Біосенсор фактично реагує на концентрацію НАДГ, однак швидкість реакції залежить від концентрації\(\text{NH}_4^+\). Якщо початкові концентрації 2-оксиглутарата і НАДГ однакові для всіх зразків і стандартів, то сигнал пропорційний концентрації\(\text{NH}_4^+\). Як показано в наступній таблиці, чутливість методу залежить від рН.
| рН | чутливість (нА S —1 М —1) |
|---|---|
| 6.2 | \(1.67 \times 10^3\) |
| 6.75 | \(5.00 \times 10^3\) |
| 7.3 | \(9.33 \times 10^3\) |
| 7.7 | \(1.04 \times 10^4\) |
| 8.3 | \(1.27 \times 10^4\) |
| 9.3 | \(2.67 \times 10^3\) |
Двома можливими поясненнями впливу рН на чутливість цього аналізу є кислотно-основна хімія ферменту\(\text{NH}_4^+\) та кислотно-основна хімія ферменту. Враховуючи, що p K a for\(\text{NH}_4^+\) дорівнює 9.244, поясніть джерело цієї pH-залежної чутливості.
31. Схема видоутворення мікрометалів у таблиці 11.4.2 ділить їх на сім операційно визначених груп шляхом збору та аналізу двох зразків після кожної з чотирьох обробок, що вимагає загалом восьми зразків та восьми вимірювань. Після видалення нерозчинних частинок шляхом фільтрації (обробки 1) розчин аналізують на концентрацію лабільних металів ASV і на загальну концентрацію металів. Потім порцію відфільтрованого розчину пропускають через іонообмінну колону (обробка 2) і визначають концентрації металу ASV і загального металу. Другу порцію відфільтрованого розчину опромінюють ультрафіолетовим світлом (обробка 3), вимірюють концентрації металу ASV і загального металу. Нарешті, третю порцію відфільтрованого розчину опромінюють ультрафіолетовим світлом і пропускають через іонообмінну колону (обробка 4), і визначають концентрації лабільного металу ASV і загального металу знову. Групи, які включені в кожне вимірювання, зведені в наступній таблиці.
| лікування | групи, видалені шляхом лікування | групи, що сприяють ASV-лабільним металам | групи, що сприяють загальному обсязі металів |
|---|---|---|---|
| 1 | жоден | I, II, ІII | I, II, III, IV, V, VI, VII |
| 2 | I, IV, V | ІІ, ІІІ | ІІ, ІІ, V1, VII |
| 3 | жоден | I, II, III, IV, VI | I, II, III, IV, V, VI, VII |
| 4 | I, II, IV, V, VI | III | ІІІ, VII |
(a) Поясніть, як можна використовувати ці вісім вимірювань для визначення концентрації металів, присутніх у кожній з семи груп, визначених у таблиці 11.4.2.
(б) Бетлі та Флоренс повідомляють про наступні результати видоутворення кадмію, свинцю та міді у зразку морської води [Батлі, Г. Е.; Флоренція, Т. М. Летт. 1976, 9, 379—388]. Визначте видоутворення кожного металу в коментарі до ваших результатів.
|
вимірювання лікування: ASV-лабільний або загальний |
ППБ Кд 2+ | ППБ 2+ | ППБ Cu 2+ |
|---|---|---|---|
| 1: ASV-лабільний | 0,24 | 0,39 | 0,26 |
| 2: всього | 0,28 | 0,50 | 0,40 |
| 2: ASV-лабільний | 0,21 | 0,33 | 0,17 |
| 2: всього | 0,26 | 0,43 | 0,24 |
| 3: ASV-лабільний | 0,26 | 0,37 | 0,33 |
| 3: всього | 0,28 | 0.5 | 0,43 |
| 4: ASV-лабільний | 0.00 | 0.00 | 0.00 |
| 4: всього | 0,02 | 0,12 | 0,10 |
32. Концентрація Cu 2+ в морській воді визначається анодною зачисткою вольтамметрії на висячому ртутному крапельному електроді після першого вивільнення будь-якої міді, пов'язаної з органічною речовиною. До 20,00-мл проби морської води додають 1 мл 0,05 М HNO 3 і 1 мл 0,1% H 2 O 2. Зразок опромінюють ультрафіолетовим світлом протягом 8 годин, а потім розводять до об'єму в об'ємній колбі об'ємом 25 мл. Осадження Cu 2 + відбувається при —0,3 В проти СКВ протягом 10 хв, виробляючи піковий струм 26,1 (довільні одиниці). Другий 20,00-мл проби морської води обробляють однаково, за винятком того, що додають 0,1 мл 5,00 мкм розчину Cu 2 +, виробляючи піковий струм 38,4 (довільні одиниці). Повідомити про концентрацію Cu 2 + в морській воді в мг/л.
33. Тіоамідні препарати визначаються за допомогою катодного зачистки аналізу [Davidson, I.E.; Smyth, WF Anal. Хім. 1977, 49, 1195—1198]. Осадження відбувається при +0,05 В проти SCE. На етапі зачистки потенціал сканується катодно і спостерігається пік зачистки при -0,52 В. При типовому застосуванні 2,00 мл проби сечі змішують з 2,00 мл буфера рН 4,78. Після осадження 2.00 хв вимірюється піковий струм 0,562 мкА. В цей же розчин додають 0,10-мл додавання 5,00 мкм розчину препарату. Піковий струм 0,837 мкА реєструється з використанням тих же умов осадження і зачистки. Повідомте про молярну концентрацію препарату в зразку сечі.
34. Концентрацію ванадію (V) в морській воді визначають методом адсорбційної стріппинг-вольтамметрії після утворення комплексу з катехолом [van der Berg, C. m.G.; Huang, Z. Q. анал. Хім. 1984, 56, 2383—2386]. Комплекс катехол-V (V) осаджується на висячому ртутному електроді з потенціалом —0,1 В проти опорного електрода Ag/AgCl. Сканування катодного потенціалу дає пік зачистки, пропорційний концентрації V (V). Наступні стандартні доповнення використовуються для аналізу зразка морської води.
| [V (V)] додано (М) | піковий струм (мкА) |
|---|---|
|
\(2.0 \times 10^{-8}\) |
24 |
| \(4.0 \times 10^{-8}\) | 33 |
| \(8.0 \times 10^{-8}\) | 52 |
| \(1.2 \times 10^{-7}\) | 69 |
| \(1.8 \times 10^{-7}\) | 97 |
| \(2.8 \times 10^{-7}\) | 140 |
Визначте молярну концентрацію V (V) у зразку морської води, припускаючи, що стандартні доповнення призводять до незначної зміни обсягу зразка.
35. Потенціал зниження стандартного стану для Cu 2 + до Cu становить +0,342 В проти SHE. Враховуючи, що Cu 2+ утворює дуже стабільний комплекс з лігандом ЕДТА, чи очікуєте ви, що потенціал відновлення стандартного стану для Cu (EDTA) 2— більше +0,342 В, менше +0,342 В, або дорівнює +0,342 В? Поясніть свої міркування.
36. Полярографічні напівхвильові потенціали (проти SCE) для Pb 2+ і для Tl + в 1 М HCl складають відповідно —0,44 В і —0,45 В в електроліті 1 М NaOH, однак напівхвильові потенціали становлять —0,76 В для Pb 2 + і —0,48 В для Tl +. Чому зміна електроліту робить такий істотний вплив на напівхвильовий потенціал для Pb 2 +, але не на напівхвильовий потенціал для Tl +?
37. Наступні дані для зниження Pb 2 + були зібрані методом нормально-імпульсної полярографії.
| потенціал (V проти SCE) | струм (мкА) |
|---|---|
| —0,345 | 0,16 |
| —0,370 | 0,98 |
| —0,383 | 2.05 |
| —0,393 | 3.13 |
| —0,409 | 4.62 |
| —0,420 | 5.16 |
Граничний струм становив 5,67 мкА. Переконайтеся, що реакція відновлення оборотна, і визначте значення для n і E 1/2. Півхвильові потенціали для нормально-імпульсних полярограм Pb 2+ при наявності декількох різних концентрацій ОН — наведені в наступній таблиці.
| [О —] (М) | \(E_{1/2}\)(V проти SCE) | [О —] (М) | \(E_{1/2}\)(V проти SCE) |
|---|---|---|---|
| 0,050 | \ (E_ {1/2}\) (V проти ПКЄ) ">—0.646 | 0,150 | \ (E_ {1/2}\) (V проти ПКЄ) ">—0.689 |
| 0.100 | \ (E_ {1/2}\) (V проти ПКЄ) ">—0.673 | 0,300 | \ (E_ {1/2}\) (V проти ПКЄ) ">—0.715 |
Визначають стехіометрію комплексу РБ-гідроксиду і константу його утворення.
38. У 1977 році, коли я був студентом в коледжі Нокс, мій партнер лабораторії і я завершили експеримент з вивчення вольтамметричної поведінки Cd 2 + (в 0.1 M KNO 3) і Ni 2 + (в 0.2 M KNO 3) при падаючому ртутному електроді. Дані в цій проблемі взяті з мого лабораторного ноутбука. Всі потенціали відносяться до опорного електрода SCE.
| потенціал для Cd 2+ (V) | струм для Cd 2+ (мкА) | потенціал для Ni 2+ (V) | струм для Ni 2+ (мкА) |
|---|---|---|---|
| —0,60 | 4.5 | —1.07 | 1,90 |
| —0,58 | 3.4 | —1.05 | 1.75 |
| —0,56 | 2.1 | —1.03 | 1,50 |
| —0,54 | 0.6 | —1.02 | 1,25 |
| —0,52 | 0.2 | —1,00 | 1.00 |
Граничні струми для Cd 2 + становили 4,8 мкА, а для Ni 2 + - 2,0 мкА. Оцініть електрохімічну оборотність для кожного іона металу і прокоментуйте свої результати.
39. Болдуін та колеги повідомляють наступні дані циклічного дослідження вольтамметрії електрохімічної поведінки р -фенілендіаміну в буфері рН 7 [Болдуін, Р. П.; Ravichandran, K.; Johnson, R.K. J. Chem. Едук. 1984, 61, 820—823]. Всі потенціали вимірюються відносно SCE.
| швидкість сканування (мВ/с) | Е р, а (В) | Е р, с (В) | i р, (мА) | i р, с (мА) |
| 2 | 0,148 | 0,104 | 0,34 | 0,30 |
| 5 | 0.149 | 0.098 | 0,56 | 0,53 |
| 10 | 0.152 | 0.095 | 1.00 | 0,04 |
| 20 | 0.161 | 0.095 | 1.44 | 1.44 |
| 50 | 0.167 | 0.082 | 2.12 | 1.81 |
| 100 | 0,180 | 0.063 | 2.50 | 2.19 |
Початкове сканування спрямоване на більше позитивних потенціалів, що призводить до реакції окислення, показаної тут.
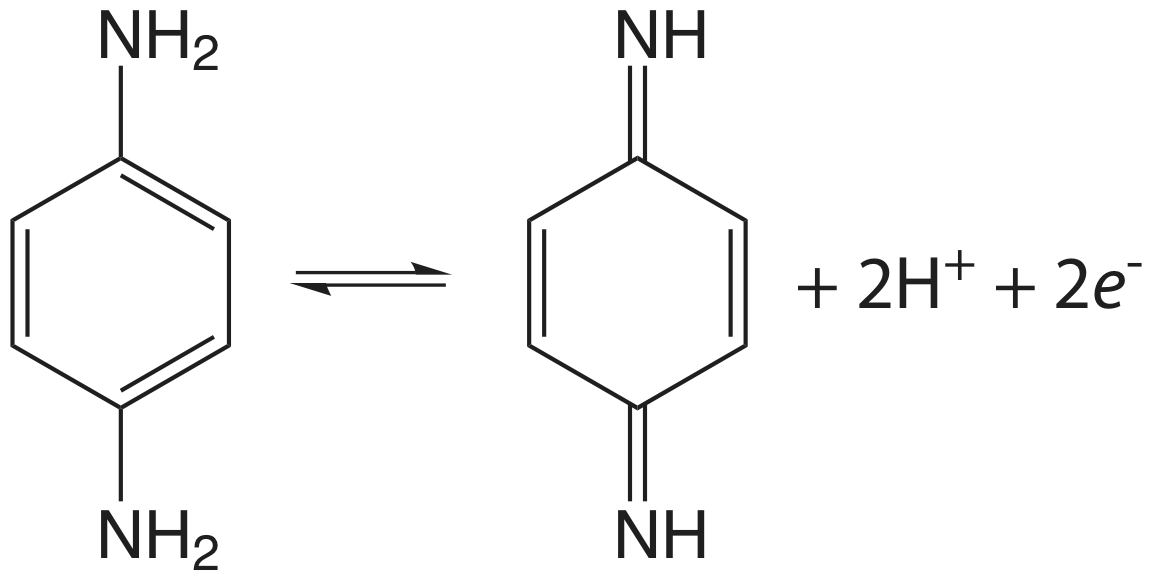
Використовуйте ці дані, щоб показати, що реакція електрохімічно незворотна. Реакція може проявляти електрохімічну незворотність через повільну кінетику перенесення електронів або через те, що продукт реакції окислення бере участь у хімічній реакції, яка виробляє неелектроактивний вид. Виходячи з даних цієї проблеми, що є ймовірним джерелом електрохімічної незворотності р -фенілендіаміну?