5.12: Піросеквенування
- Page ID
- 5765

У лабораторіях по всьому світу виникає інтенсивне бажання послідовності більшої кількості геномів.
- ті, що мають найрізноманітніші організми, щоб допомогти у встановленні еволюційних відносин;
- групи об'єднаних популяцій мікроорганізмів у, наприклад, морській воді, грунті, товстому кишечнику;
- інші люди шукати гени, які привертають до хвороб та генетичних закономірностей у різних етнічних групах.
Всі секвеновані геноми, перераховані в розмірах геному, були визначені за допомогою методу дідеокси, винайденого Фредеріком Сангером і описаного в іншому місці. Однак зараз витрачаються великі зусилля, щоб знайти способи послідовності ДНК швидше (і дешевше).
Секвенсор геному
Розробляється кілька нових методів, і один вже комерційно доступний (система Genome Sequencer 20). Його метод називається піросеквенування або секвенування шляхом синтезу. Це працює так.
- ДНК, що підлягає секвенуванню, розбивається на фрагменти ~ 100 пар основ і денатурується з утворенням одноцепочечной ДНК (SSDNA).
- Одиночні фрагменти SSDNA прикріплені до мікроскопічних кульок, які відокремлені один від одного.
- Полімеразна ланцюгова реакція (ПЛР) виконується на кожній кульці так, що кожен покривається ~ 10 мільйонами однакових копій цього фрагмента.
- Намистини поміщаються поодинці в окремі мікроскопічні лунки (з них ~ 200 000).
- Кожна лунка отримує коктейль з реактивів:
- ДНК-полімераза — для додавання дезоксирибонуклеотидів до SSDNA
- аденозинфосфосульфат (APS)
- АТФ сульфурилаза — фермент, який утворює АТФ з аденозинфосфосульфату (АПС) і пірофосфату (PPi)
- люциферин
- люцифераза - АТФаза, яка каталізує перетворення люциферину в оксилюциферин з вивільненням світла
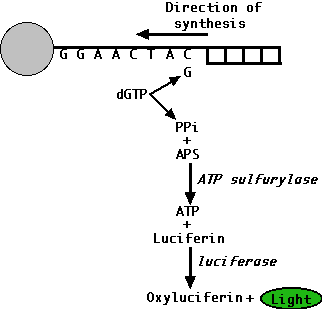

Послідовність виконання:
- Кожна з тисяч свердловин затоплена одним чотирма дезоксирибонуклеотидами, dTTP, DctP та dGTP, але замість DATP (який би викликав реакцію люциферину) замість дезоксиаденозин-альфа-тіотрифосфат (DATPαs). ДНК-полімераза ігнорує різницю і використовує її щоразу, коли на шаблоні SSDNA зустрічається T, але люцифераза її не розпізнає.
- У будь-якій лунці, де додатковий нуклеотид присутній на 3' кінці шаблону, нуклеотид додається і пірофосфат звільняється.
- Кількість світла пропорційна кількості доданого нуклеотиду. Так що якщо, наприклад, вхідний нуклеотид dGTP, а на шаблоні є рядок 3 Cs, випромінюване світло буде в 3 рази яскравіше, ніж якщо присутній тільки один С.
- Детектор забирає світло (якщо є) з кожної свердловини, і дані записуються.
- Потім кожен з інших 3 нуклеотидів додається послідовно.
- Потім послідовність з 4 додавань повторюється до завершення синтезу.
На наведеній вище схемі також показаний тип даних, вироблених в одній свердловині. Висота піку світлового виробництва дає кількість доповнень, які виникли при додаванні певного нуклеотиду (знизу). Комп'ютерне програмне забезпечення потім відображає послідовність шаблонів (зверху) для кожного з тисяч різних фрагментів, послідовних. За допомогою цієї технології, цілих 20 мільйонів пар основ послідовності генома можна дізнатися за допомогою інструменту менше 6 годин.