8.11: Послідовність наступного покоління
- Page ID
- 6512

Розширення технології секвенування
Кредит: Джеремі Сето (CC-BY-NC-SA 3.0)
Традиційне секвенування геномів було тривалим і виснажливим процесом, який клонував фрагменти геномної ДНК в плазміди для створення геномної бібліотеки ДНК (gDNA). Ці плазміди були індивідуально секвеновані за допомогою методології секвенування Сангера, і обчислювальні були виконані для ідентифікації частинок, що перекриваються, як головоломки. Ця збірка призведе до проекту риштування.
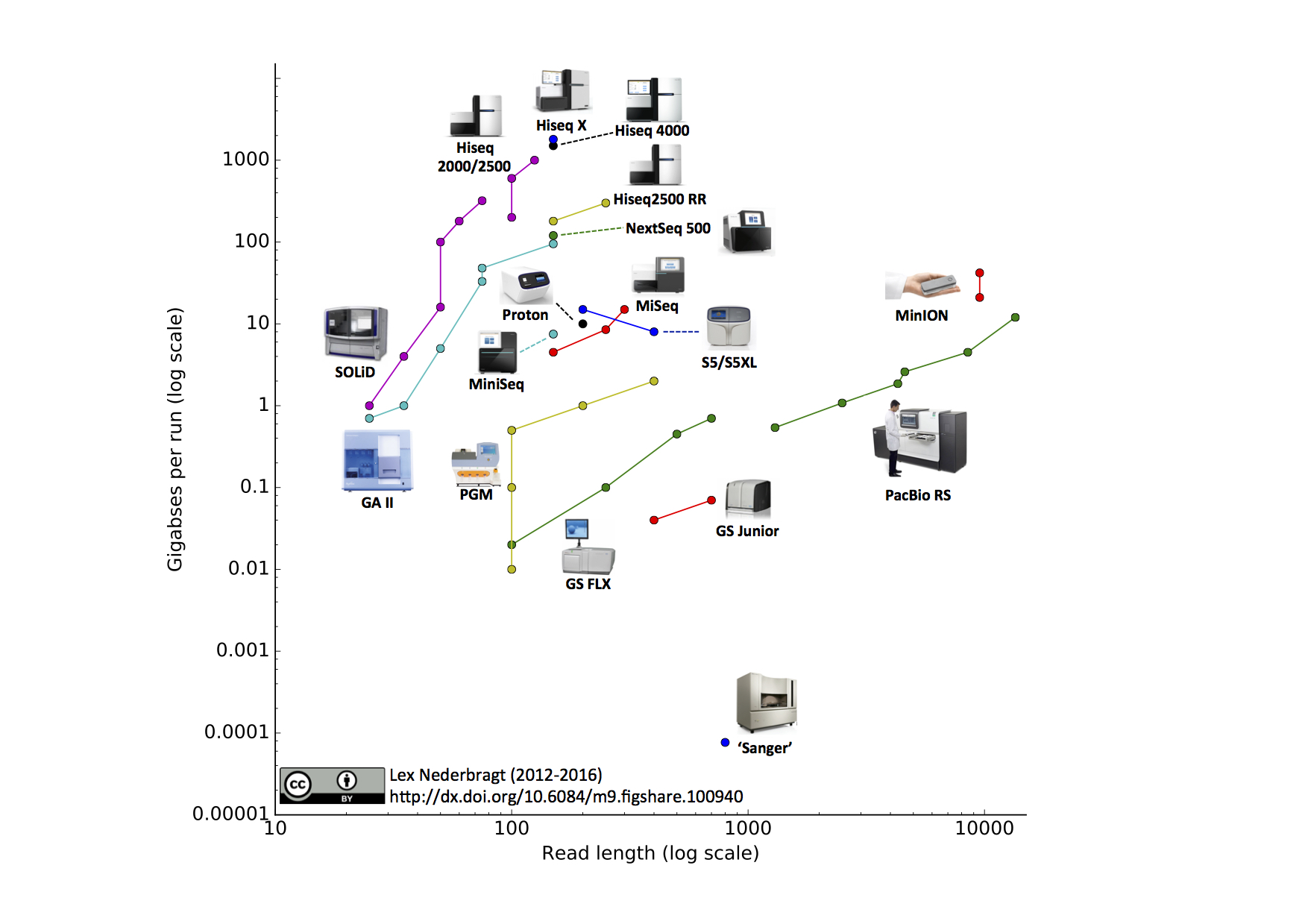
У міру вдосконалення технології вартість секвенування геномів стала менш дорогою. Ця технологія випередила закон Мура, напівпровідникову проекцію про швидкість комп'ютерів з плином часу. Різке зниження ціни на секвенування геному відбулося приблизно в 2008 році через технічні досягнення.

Зі зниженням вартості секвенування генома спостерігалося різке збільшення осадження геному в Генбанк. Ці поклади відображали дрібні геноми бактерій і архей.

Кредит: Естевеж (CC-BY-SA 3.0)
Зниження вартості секвенування на нуклеотид відбулося внаслідок паралелізації секвенування. У той час як Секвенування Sanger здатне секвенувати один розтяг за раз, паралельна збірка реакцій секвенування призвела до високої пропускної здатності секвенування, яке часто називають секвенуванням наступного покоління (NGS).
Секвенування з високою пропускною здатністю, застосоване для секвенування геному (TEDEd CC BY-NC-ND 4.0)
Коротке читання секвенування за допомогою синтезу
Ілюміна
Послідовність короткого читання Illumina використовує технологію потокових клітин, де олігонуклеотиди, що доповнюють адаптерні грунтовки, фізично висівають.

Проточні поверхні клітин з перехідником олігонуклеотидів. Кредит: ДМлапато (CC-BY-SA 4.0)
Фрагментовані послідовності ДНК адаптовані за допомогою праймерів шляхом лігування та гібридизуються до проточної клітини. Щоб збільшити сигнал від секвенування, короткі послідовності ДНК посилюються за допомогою процесу, який називається ампліфікацією мостів або генерацією кластерів.

Генерація кластерів за допомогою посилення мостів. Застосовується низький онімілий ер циклів ПЛР. Кластерна генерація допомагає в подальшому визначенні сигналу/шуму.
Кредит: ДМлапато (CC-BY-SA 4.0)
Проточна клітина проходить послідовні раунди затоплення флуоресцентним нуклеотидом, дозволеним до включення з ДНК-полімеразою і змивається. Після кожного циклу повень/прання вимірюються флуоресцентні сигнали для позначення включення. Конкретні місця флуоресценції відстежуються та консолідуються для позначення послідовності в кожній зареєстрованій точці.

Кожен цикл потоку вводить флуоресцентний нуклеотид для включення. Кредит: Абізар Лакдавалла (CC-BY 3.0)
Іонний торрент
Фрагментована ДНК перев'язана до послідовностей адаптерів і приклеюється до мікрокульок. Намистини вбудовані в мікросвердловини на напівпровіднику. Ion Torrent виконує реакції секвенування в небуферизованому розчині, оскільки напівпровідник діє як рН-метр для ідентифікації включення нуклеотидів. Стандартні нуклеотиди заливаються на чіп і вбудовуються. Оскільки включення нуклеотидів створює протон (H +), мікросередовище з низьким рН виявляється в небуферизованому розчині.
Секвенування в режимі реального часу з однією молекулою
Пак Біо
Pac Bio використовує нано-лунки з ковалентно-зв'язаною ДНК-полімеразою для послідовності окремих молекул ДНК. Флуоресцентні нуклеотиди включаються під час реакцій синтезу, і в режимі реального часу можна виміряти включення. Секвенування Pac Bio має перевагу секвенування фрагментів 10-20kb, на відміну від коротких методів.
Оксфорд — Нанопор

Кредит: Георгіївська церква (CC-BY 3.0)
Oxford Nanopore використовує білок альфа-гемолізин, інтегрований на напівпровідниковий чіп. Розмір пор білка є правильним розміром для однієї молекули ДНК, щоб пройти. Молекула ДНК-полімерази пов'язана з відкриттям пори, через яку подається реплікована ДНК. Коли ДНК проходить пори, зміни напруги вимірюються та відображаються на якості конкретних основ.
Посилання: Юту.бе/БНЗ880В52РК
Вивід послідовності
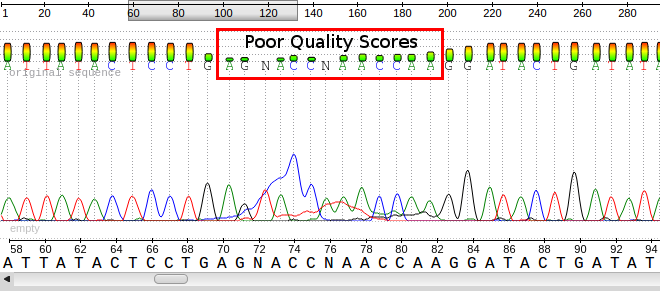
Зразок файлу ab1, що відображає базові дзвінки, хроматограми та оцінки якості для кожної бази. Зверніть увагу на погану якість у червоній коробці та відповідні піки/основи.
Вихідний файл методів послідовності наступного покоління використовує формат fastq. Як і файл fasta, є заголовок, який описує послідовність. Перший рядок - це заголовок або рядок заголовка, який починається з '@' (пам'ятайте, що fasta починається з '>'). Другий рядок - це фактична сира послідовність (ще раз схожа на фасту). Третій рядок не має ніякого значення, тоді як четвертий рядок заповнюється символами до тих пір, поки рядок послідовності. Цей останній рядок є оцінкою якості базового виклику. Як і у випадку з секвенуванням Сангера, може виникнути неоднозначність з базовим викликом послідовності, і впевненість зберігається в оцінці якості.
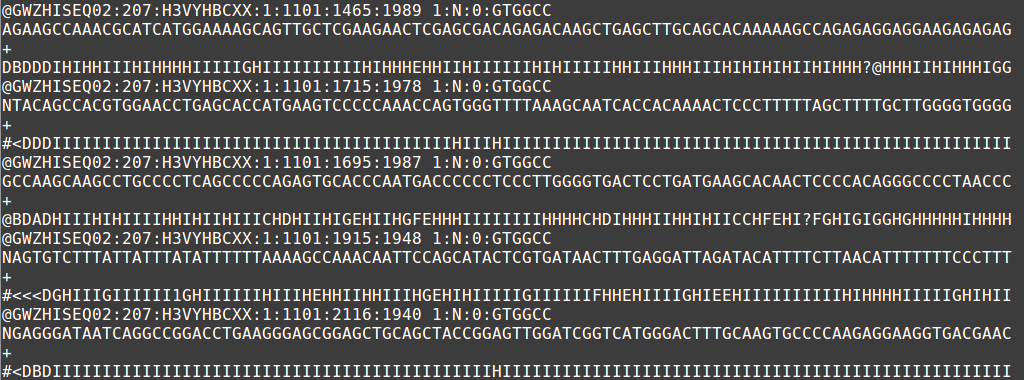
Приклад файлу fastq, що відображає 5 коротких послідовностей читання.
Оцінки Phred були розроблені для оцінки якості базових викликів, що виникають внаслідок флуоресцентного секвенування Сангера під час проекту «Геном людини». Програма Phred сканує піки хроматограми і бали на основі визначеності або точності дзвінка. Оцінки засновані на логарифмічній основі, а бали більше 20 представляють точність, що перевищує 99% базового виклику.
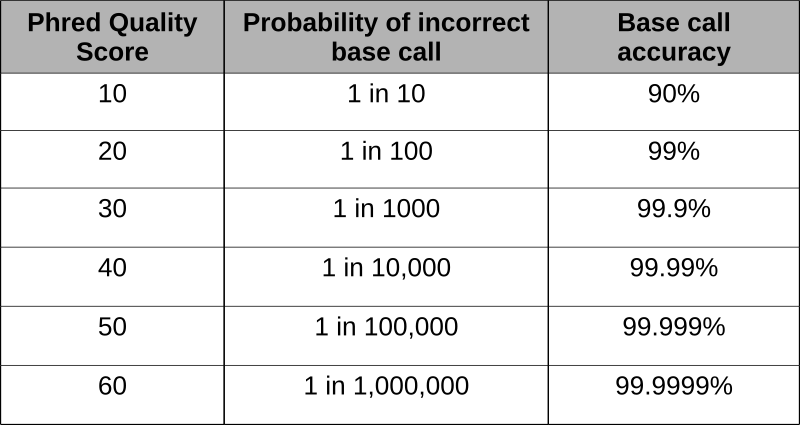
Використовуючи бали phred, вбудовані в останній рядок файлів fastq, неякісні читання можуть бути видалені. Використання такої програми, як FastQC дозволяє оцінювати читання і виробляє графічне зображення якості.
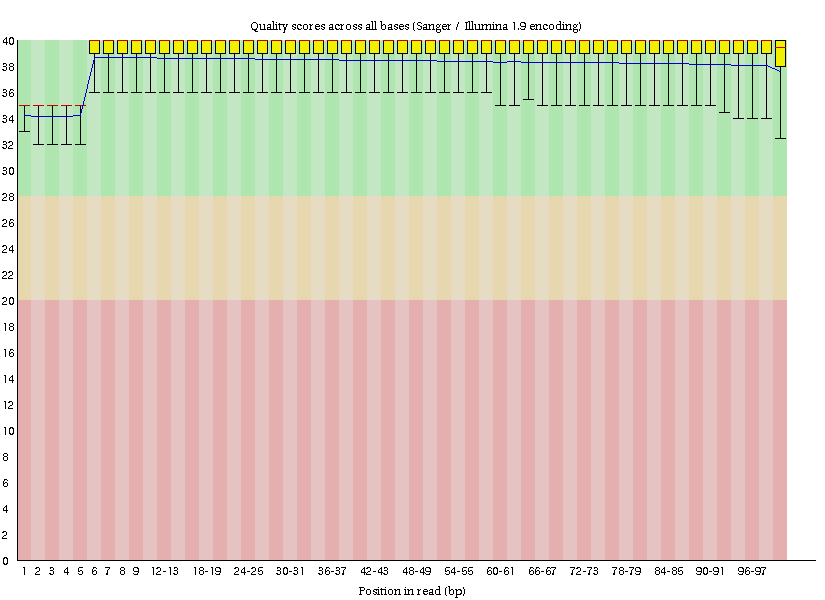
Вихід якості FastQC, що ілюструє показник Phred для кожного базового виклику.
Ця коротка послідовність читання близько 100 нуклеотидів має всі основи, зроблені з точністю понад 30, або > 99,9%.
Збірка і вирівнювання
Послідовності з коротких читань повинні бути зібрані в корисну послідовність. Для цього еталонний геном може допомогти в збірці після того, як послідовності адаптерів обрізаються автоматизованими методами. У випадку, якщо еталонного генома немає, може бути використаний споріднений вид або має відбутися більш обчислювально-інтенсивний процес складання de novo. При збірці de novo може бути корисним мати деякі довгі читання, виконані з PacBio для створення риштування для генерації збірки в суміжні послідовності, або контиги.

РНК-SEQ

Кредит: Малахії Гріффіт, Джейсон Уокер, Ніколас Шпигуни, Бенджамін Ейнсуф, Обі Л.Гріффіт (CC-BY 4.0) https://doi.org/10.1371/journal.pcbi.1004393

Кредит: Rgocs (CC-BY)
RT-PCR та RT-QPCR можуть бути використані для вимірювання кількості конкретних стенограм досить низькою пропускною здатністю. Використовуючи концепцію зворотної транскрипції та зв'язку, що з технологіями секвенування з високою пропускною здатністю, стенограми можуть бути послідовні та відображені на геномі, щоб зобразити кількість стенограм, представлених кількістю читань.
Враховуючи достатнє покриття читання, нові ізоформи зрощування також можуть бути ідентифіковані як різні екзон-екзонові переходи ідентифіковані.
Загальний робочий процес аналізу РНК-SEQ наступний:

Кредит: Цілюще токсин (CC-BY-SA 4.0)