26.1: Результати рівноваги, коли енергія Гіббса зведена до мінімуму
- Page ID
- 26720
Багато важливих хімічних реакцій - якщо не більшість - проводяться в розчині, а не між твердими речовинами або газами. Реакції твердого тіла часто дуже повільні, і не всі хімічні види можуть бути введені в парову фазу, оскільки вони розкладаються перед випаровуванням.
Часто нас не турбують тимчасові аспекти реакції. Це може бути технологічно дуже важливим, але це область кінетики - інша галузь фізичної хімії - а не класична термодинаміка.. Останній більше стосується кінцевої точки. Це термодинамічно кажучи (стабільна) рівновага, але хімічно воно може або представляти завершену реакцію, або хімічну рівновагу.
На жаль, з трьох основних агрегатних станів: газ — рідина — тверда, структура рідин найменш зрозуміла, і одна з найскладніших рідин також є однією з найбільш широко використовуваних: вода. Це життєво важливо для багатьох галузей хімії, що варіюються від геохімії до хімії навколишнього середовища до біохімії. Ми зробимо лише невелику внутрішню дорогу в її складність.
ступінь реакції
Для опису прогресу реакції ми визначаємо ступінь реакції. Зазвичай позначається грецькою літерою\(ξ\).
Розглянемо родову реакцію:
\[v_AA + v_BB \rightleftharpoons v_YY + v_ZZ \nonumber \]
Використовуючи стехіометрію, ми можемо визначити ступінь, враховуючи, як змінюється кількість родимок (або молярних кількостей) кожного виду під час реакції:
реагенти
- \(n_A= n_{A,0} - v_A ξ\)
- \(n_B= n_{B,0} - v_B ξ\)
продукти
- \(n_Y= n_{Y,0} + v_Y ξ\)
- \(n_Z= n_{Z,0} + v_Z ξ\)
Розмірність дорівнює [моль], оскільки стехіометричні коефіцієнти v i є безрозмірними цілими числами. Якщо реакція піде до завершення для одного з реагентів -граничного реагенту - n A або B =n обмеження піде до нуля. Якщо ми починаємо з\(n_{limiting}= v_{limiting}\) кротів, значення починається з 0 (без продуктів) і переходить до 1 після завершення (обмежуючи виснаження реагенту). При наближенні рівноваги не\(ξ\) вийде далі\(ξ_{eq}\).
Вимірювання
Ступінь реакції - це те, що є центральним суб'єктом кінетики реакції. Його значення зазвичай вимірюється як функція часу побічно шляхом вимірювання величини q, яка лінійно залежить від (t):
\[q( ξ ) = aξ +b \nonumber \]
Розглянемо ситуацію в крайності\(ξ=0\) і\(ξ=1\):
\[q_0= a.0+b= b \nonumber \]
\[q_1= a.1+b= a+b \nonumber \]
\[q_1-q_0= a \nonumber \]
Таким чином,\(ξ\) можна знайти з
\[\dfrac{q(t)-q0}{q_1-q_0}=\dfrac{q(t)-b}{a} \nonumber \]
Характер\(q\) може сильно варіюватися від поглинання УФ/ВІС, провідності, гравіметричних даних до калорійності.
\(q_0\)На практиці часто\(ξ=0\) важко спостерігати, оскільки потрібен час для змішування реагентів, особливо в розчині, і q 1 при = 1 може ніколи не бути досягнуто, якщо реакція перейде до рівноваги. Проте значення a і b часто можна знайти з наявних даних методом примірки.
У (рівноважної, статичної) термодинаміки ми стурбовані лише кінцевою точкою:
- \(ξ=1\): реакція доходить до завершення
- \(ξ=ξ_{eq}\): реакційний іон переходить в стан хімічної рівноваги
Термодинамічні потенціали
Як ми бачили, ми можемо записати будь-яку зміну вільної енергії Гіббса через зміни молярних кількостей видів, що беруть участь у реакції (ат\(T\),\(P\) константа) як:
\[dG =\sum \dfrac{∂G}{∂n_i} dn_i = \sum μ_idn_i \nonumber \]
де\(μ\) знаходиться термодинамічний потенціал, часто називають хімічним потенціалом при роботі з реакціями. З визначення ми можемо побачити за диференціацією, що
- \(d n_A=- v_Adξ\)
- \(d n_B=- v_Adξ\)
- \(d n_Y= v_Ydξ\)
- \(d n_Z= v_Zdξ\)
Це дозволяє уніфікувати зміни молярної кількості всіх видів в одну єдину змінну\(dξ\). Отримуємо:
\[dG = \left[ \sum -v_{i,reactants} μ_{i,reactants} + \sum+v_{i,products} μ_{j,products} \right]dξ \nonumber \]
або
\[ \left (\dfrac{∂G}{∂ξ} \right)_{T,P} = -\sum v_{i,r}μ_{i,r}+ \sum v_{i,p}μ_{j,p} \nonumber \]
Ця кількість також пишеться як:
\[ \left( \dfrac{∂G}{∂ξ} \right)_{T,P} =Δ_rG \nonumber \]
Ця величина дає зміну вільної енергії Гіббса для реакції (як написано!!) для Δ= 1 моль. (Одиниці [Дж/моль] отже). Це зміна енергії Гіббса (нахил\(G\) vs\(\xi\)), коли ступінь реакції змінюється на один моль з фіксованим складом. Рівновага результати, коли енергія Гіббса знаходиться на мінімумі щодо ступеня реакції.
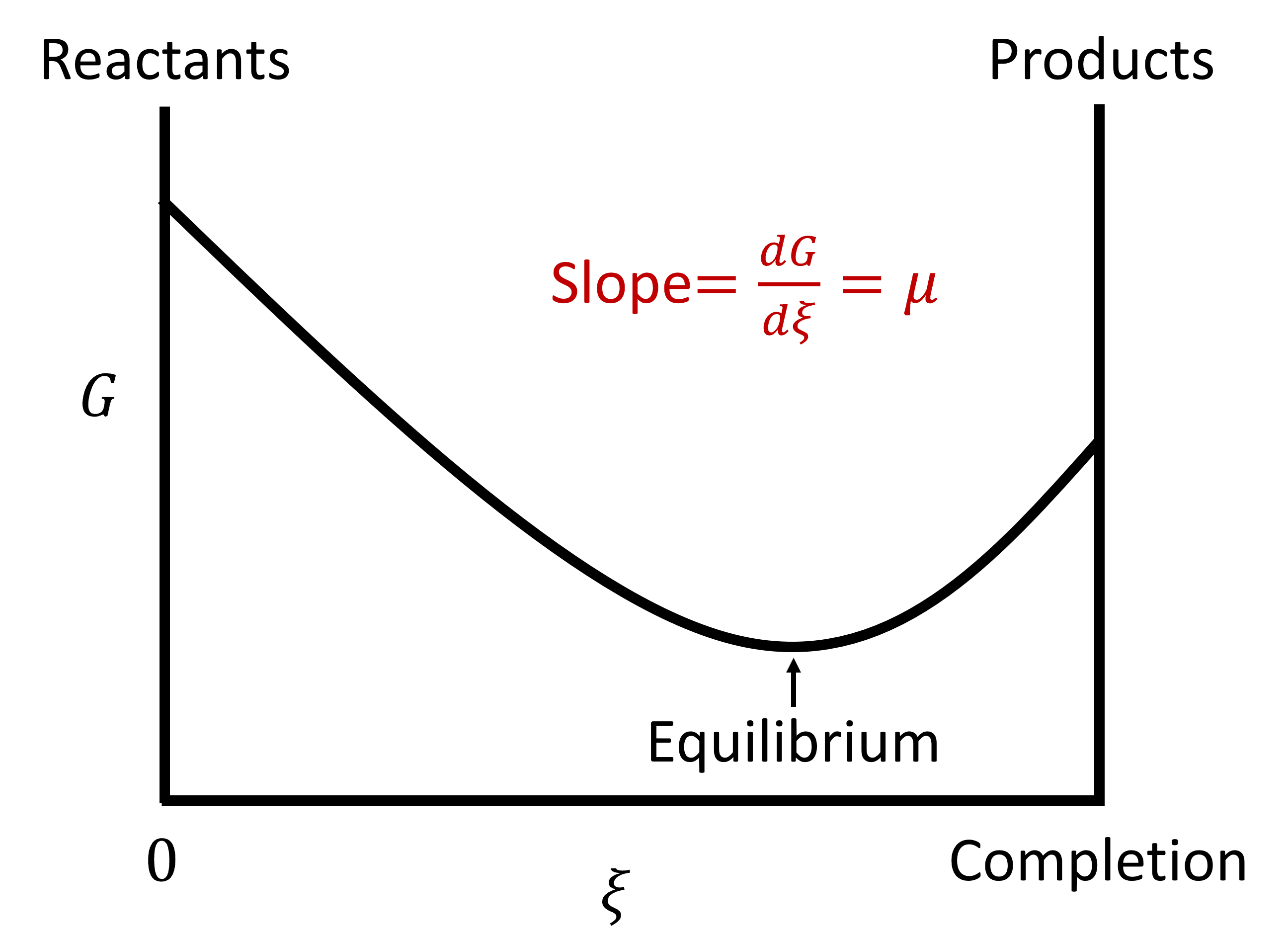
газові реакції
Припустимо, що наша реакція повністю між видами газу і що газ досить розбавлений, щоб ми могли використовувати ідеальний закон газу. Тоді ми можемо написати для кожного виду:
\[μ_i= μ_i^o+RT \ln \dfrac{P_i}{P_i^o} \nonumber \]
Потім ми можемо розділити вираз Δ r G на дві частини:
\[Δ_rG = Δ_rG^o + RT\ln Q \nonumber \]
Стандартні потенціали:
\[Δ_rG^o = -\sum v_i,_r μ^o_{i,r} + \sum v_{i,p} μ^o_{j,p} \nonumber \]
і логарифмічні терміни:
\[RT \ln Q= - v_A RT \ln \left( \dfrac{P_A}{P_A^o} \right)- v_B RT\ln \left( \dfrac{P_B}{P_B^o} \right) + v_YRT \ln \left( \dfrac{P_Y}{P_Y^o} \right) + v_ZRT \ln \left( \dfrac{P_Z}{P_Z^o} \right) \nonumber \]
Ми можемо об'єднати всі логарифмічні терміни в Q, званий коефіцієнтом реакції. Стехіометричні коефіцієнти стають показниками, а фактори реагентів будуть «догори ногами» порівняно з продуктами через властивості логарифмів:
\[a \ln x = \ln x^a \nonumber \]
\[- a \ln x = \ln \left( \dfrac{1}{x^a} \right) \nonumber \]
Ми зберегли стандартний тиск\(P_i^o\) у виразі, але часто вони опускаються. Вони, як правило, всі 1 бар, але в принципі ми могли б вибрати 1 бар для A 1 Torr для B і 1 psi для продуктів. Це створює дійсне (хоча і смішне) визначення того, що означає o. (Звичайно, значення\(Δ_rG^o\) дійсно залежить від цього вибору!).
Ми могли б написати
\[RT \ln Q = RT \ln \dfrac{Q_P}{Q^o} \nonumber \]
\(Q^o\)як правило, єдність за величиною, але він скасовує розміри\(Q_P\). Це означає,\(Q_P\) що\(Q\) і рівні за величиною, і ми можемо отримати\(Q\), просто скинувши розміри.\(Q_P\) \(Q\)безрозмірний, але\(Q_P\) зазвичай це не так. Часто це прекрасне розмежування просто не\(Q^o\) проводиться і опускається, отримуємо:
\[Δ_rG = Δ_rG^o + RT\ln \dfrac{P_Y^{v_Y}P_Z^{v_Z} }{P_A^{v_A}P_B^{v_B}} \nonumber \]
Зверніть увагу на різницю між\(Δ_rG\) якими позначає умови (наприклад, тиск) вашої реакції та\(Δ_rG^o\) позначає стандартні умови.