2.2: Побудова молекулярних орбіталів з атомних орбіталів
- Page ID
- 19954

Молекулярна орбітальна теорія передбачає розв'язування (приблизно) рівняння Шредінгера для електронів в молекулі. Для перегляду з глави 1, це диференціальне рівняння, в якому перший і другий члени праворуч представляють кінетичну та потенційну енергії:
\[E \psi = -\frac{\hbar^{2}}{2\mu} \nabla^{2} \psi + V \psi\]
Хоча рівняння Шредінгера може бути вирішено аналітично для атома водню, функція потенційної енергії V ускладнюється - і рівняння може бути вирішено лише чисельно - коли в молекулі багато (взаємно відштовхуючих) електронів. Отже, в якості першого наближення ми будемо вважати, що s, p, d, f тощо орбіталі атомів, що складають молекулу, є хорошими розв'язками рівняння Шредінгера. Потім ми можемо дозволити цим хвильовим функціям втручатися конструктивно і руйнівно, коли ми об'єднуємо атоми, щоб створити зв'язки. Таким чином, ми використовуємо атомні орбіталі (AO) як основу для побудови МО.
LCAO-MO = лінійна комбінація атомних орбіталей. У фізиці це називається наближенням жорсткої прив'язки.
Ми насправді бачили лінійні комбінації атомних орбіталів раніше, коли ми будували гібридні орбіталі в главі 1. Тут також застосовуються основні правила, які ми розробили для гібридизації: орбіталі додаються скалярними коефіцієнтами (c) таким чином, щоб отримані орбіталі були ортогональними і нормалізованими. Різниця полягає в тому, що в випадку МО атомні орбіталі походять від різних атомів.
Лінійна комбінація атомних орбіталей завжди дає однакову кількість молекулярних орбіталей. Так що, якщо ми почнемо з двох атомних орбіталів (наприклад, s і p z орбітальний, як показано на рис. \(\PageIndex{1}\)), ми закінчуємо двома молекулярними орбіталями. Коли атомні орбіталі додають фазу, ми отримуємо конструктивну інтерференцію і нижчу енергію орбіталі. Коли вони складаються з фази, ми отримуємо вузол, і отримана орбіталь має більш високу енергію. Менші енергії MoS є склеюванням, а більш висока енергія MoS є антизв'язуючими.
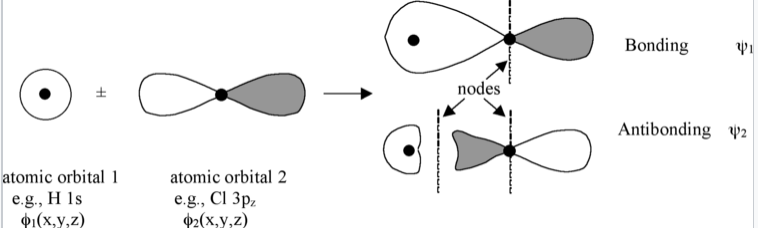
Молекулярні орбіталі також називають хвильовими функціями (ψ), оскільки вони є розв'язками рівняння Шредінгера для молекули. Атомні орбіталі (також звані базисними функціями) позначені як φ, наприклад, φ 1s і φ 3pz або просто як φ 1 і φ 2.
В принципі, нам потрібно вирішити рівняння Шредінгера для всіх орбіталів молекули, а потім заповнити їх парами електронів, як ми робимо для орбіталів в атомах. На практиці нас дійсно цікавлять лише МО, які походять від валентних орбіталів складових атомів, оскільки це орбіталі, які беруть участь у зв'язуванні. Нас особливо цікавлять прикордонні орбіталі, тобто найвища зайнята молекулярна орбіталь (HOMO) та найнижча незайнята молекулярна орбіталь (LUMO). Заповнені орбіталі, які набагато нижчі за енергією (тобто серцевинні орбіталі), не сприяють склеюванню, а порожні орбіталі при вищій енергії також не сприяють. Ці орбіталі, однак, важливі у фотохімії та спектроскопії, які включають електронні переходи від зайнятих до порожніх орбіталів. Флуоресцентні барвники, які забарвлюють клітини, показані на рис. \(\PageIndex{2}\)поглинають світло, просуваючи електрони в HOMO, щоб спорожнити MoS і видавати світло, коли електрони падають назад до початкових енергетичних рівнів.
Як приклад підходу LCAO-MO ми можемо побудувати два МО (ψ 1 і ψ 2) молекули HCl з двох AO φ 1 і φ 2 (рис.2.1.1). Щоб скласти ці дві лінійні комбінації, пишемо:
\[\Psi_{1}=c_{1}\varphi_{1} + c_{2}\varphi_{2}\]
і
\[\Psi_{2}=c_{1}\varphi_{1} - c_{2}\varphi_{2}\]
Коефіцієнти c 1 і c 2 будуть рівними (або майже так), коли два AoS, з яких вони побудовані, однакові, наприклад, коли дві водневі 1s орбіталі об'єднуються, щоб зробити склеювання та антизв'язування MoS в H 2. Вони будуть нерівні, коли існує різниця в енергії між AoS, наприклад, коли воднева орбітальна 1s та орбітальна хлор 3p об'єднуються, щоб зробити полярний зв'язок H-Cl.
Вузли:
Хвильові функції φ і ψ - це амплітуди, які пов'язані з ймовірністю знаходження електрона в якійсь точці простору. Вони мають частки з (+) або (-) знаками, які ми вказуємо затіненням або кольором. Скрізь, де хвильова функція змінює знак, у нас є вузол. Як видно на рис. \(\PageIndex{1}\), вузли в MoS виникають внаслідок руйнівної інтерференції (+) і (-) хвильових функцій. Як правило, чим більше вузлів, тим вище енергія орбіти.
У наведеному вище прикладі ми намалювали спрощену картину орбіталі Cl 3p z і отриманого MoS, залишаючи поза радіальним вузлом. Нагадаємо, що 2p орбіталі не мають радіальних вузлів, 3p орбіталі мають один, як показано на рис. \(\PageIndex{3}\). 4p орбіталі мають два радіальних вузла і так далі. MO, які ми робимо шляхом об'єднання AoS, також мають ці вузли.

Нормалізація:
Ми квадратично хвильові функції, щоб отримати ймовірності, які завжди позитивні або нульові. Отже, якщо електрон знаходиться в орбіті φ 1, ймовірність знаходження його в точці xyz дорівнює квадрату [1] φ 1 (x, y, z). Загальна ймовірність не змінюється, коли ми об'єднаємо АО, щоб зробити MoS, тому для простого випадку об'єднання φ 1 і φ 2 зробити ψ 1 і ψ 2,
\[\Psi_{1}^{2} + \Psi_{2}^{2} = \varphi_{1}^{2} + \varphi_{2}^{2}\]
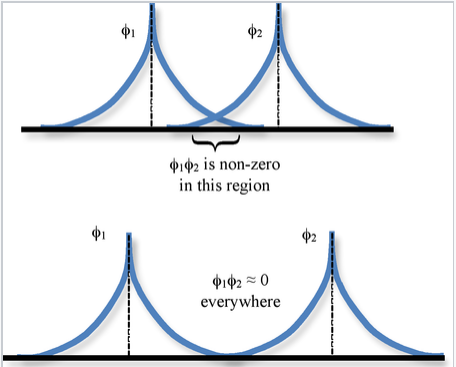
Інтегральне перекриття:
Просторове перекриття між двома атомними орбіталями φ 1 і φ 2 описується інтегралом перекриття S,
\[S_{12} = \int \varphi_{1} * \varphi_{2} d\tau \]
де інтеграція відбувається по всьому простору\(d\tau = dx dy dz\).
Енергії склеювання та антизв'язування MoS:
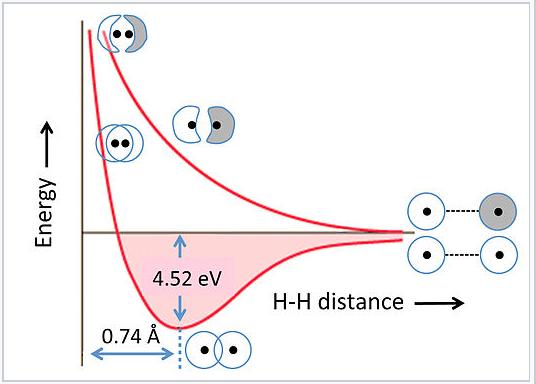
Енергії зв'язку і антизв'язуючих орбіталів сильно залежать від відстані між атомами. Це проілюстровано на рис. 2.1.5 для молекули водню, Н 2. На дуже великих відстанях суттєво відсутня різниця в енергії між інфазовими та позафазовими комбінаціями орбіталей H 1s. Коли вони наближаються, комбінація в фазі (зв'язування) падає в енергії, оскільки електрони діляться між двома позитивно зарядженими ядрами. Енергія досягає мінімуму на відстані рівноважного зв'язку (0.74 Å), а потім знову піднімається, коли ядра зближуються. Комбінація антизв'язування має вузол між ядрами, тому її енергія постійно зростає, коли атоми зводяться.
На рівноважній відстані зв'язку енергії сполучної і антизв'язуючої молекулярних орбіталей (ψ 1, ψ 2) відповідно нижче і вище енергій орбіталів атомної основи φ 1 і φ 2. Це показано на рис. \(\PageIndex{6}\)для МО молекули Н 2.
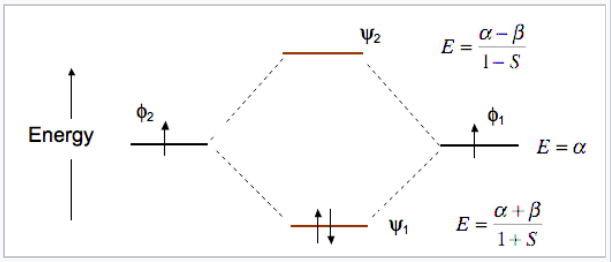
Енергія електрона в одній з атомних орбіталей дорівнює α, кулонівському інтегралу.
\[\alpha = \int \varphi_{1} H \varphi_{1}d\tau\]
де H - гамільтоновий оператор. По суті, α являє собою енергію іонізації електрона в атомній орбіталі φ 1 або φ 2.
Різниця енергій між електроном в АТ і МО визначається обмінним інтегралом β,
\[\beta = \int \varphi_{1}H\varphi_{2}d\tau\]
β є важливою величиною, тому що вона говорить нам про енергію зв'язку молекули, а також про різницю в енергії між зв'язуючими і антизв'язуючими орбіталями. Обчислення β не є простим для багатоелектронних молекул, оскільки ми не можемо розв'язати рівняння Шредінгера аналітично для хвильових функцій. Однак ми можемо зробити деякі наближення для чисельного обчислення енергій та хвильових функцій. У наближенні Гюкеля, яке може бути використано для отримання наближених розв'язків для π молекулярних орбіталей в органічних молекулах, ми спрощуємо математику, взявши S=0 і встановивши H=0 для будь-яких p-орбіталей, які не сусідять один з одним. Розширений метод Хюкеля, [2] розроблений Роальдом Гофманом, та інші напівемпіричні методи можуть бути використані для швидкого отримання відносних орбітальних енергій, наближені хвильові функції та виродження молекулярних орбіталів для широкого спектру молекул і розширених твердих тіл. Більш складні методи ab initio тепер легко доступні в програмних пакетах і можуть бути використані для обчислення точних орбітальних енергій для молекул і твердих тіл.
Отримати коефіцієнти c 1 і c 2 для молекули водню можна, застосувавши критерій нормалізації:
\[\Phi_{1} = (\varphi_{1} + \varphi_{2})/(\sqrt{2(1+S)}) \textrm{(bonding orbital)}\]
і
\[\Phi_{2} = (\varphi_{1} - \varphi_{2})/(\sqrt{2(1-S)}) \textrm{(antibonding orbital)}\]
У випадку, коли S≈0, ми можемо усунути члени 1-S, і обидва коефіцієнти стають 1/√2
Зверніть увагу, що орбітальна зв'язок на діаграмі МО Н 2 стабілізується енергією β/1+S, а антизв'язуюча орбітальна дестабілізується β/1-S. Тобто антібондинговая орбіта піднімається в енергії більше, ніж орбітальна зв'язок опускається вниз. Це означає, що H 2 (ψ 1 2 ψ 2 0) енергетично більш стійкий, ніж два атоми Н, але He 2 з чотирма електронами (ψ 1 2 ψ 2 2 2) нестійкий щодо двох атомів He.
Порядок зв'язку: На будь-якій діаграмі МО порядок зв'язку можна обчислити як ½ (# зв'язкових електронів - # антизв'язкових електронів). Для H 2 порядок облігацій дорівнює 1, а для He 2 порядок облігацій дорівнює нулю.
Гетеронуклеарний випадок (наприклад, HCl) - Полярні зв'язки
Тут ми вводимо різницю електронегативності між двома атомами, що утворюють хімічний зв'язок. Енергія електрона в орбіталі H 1s вище (її легше іонізувати), ніж електрона в орбіталі хлору 3p z. Це призводить до більшої різниці енергій між одержаними молекулярними орбіталями ψ 1 і ψ 2, як показано на рис. \(\PageIndex{7}\). Чим більша різниця електронегативності між атомними орбіталями (чим більше Δα), тим більше «φ 2 характер» має орбіталь зв'язку, тобто тим більше вона нагадує орбіталь Cl 3p z в цьому випадку. Це узгоджується з ідеєю, що H-Cl має полярну єдину зв'язок: два електрони знаходяться в зв'язковій молекулярній орбіталі, яка в першу чергу локалізується на атомі Cl.
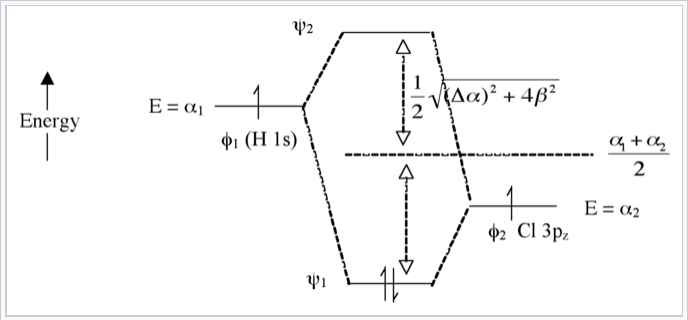
Антизв'язуюча орбітальна (порожня) має більше Н-характер. Порядок зв'язку знову дорівнює 1, тому що в орбіталі зв'язку є два електрони і жоден в антизв'язуючій орбіті.
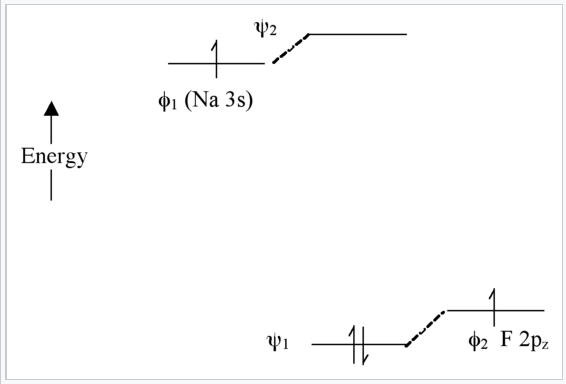
Крайній випадок - Іонний зв'язок (NaF): дуже великий Δα
У цьому випадку між АТ не так багато змішування, оскільки їх енергії знаходяться далеко один від одного (рис. \(\PageIndex{8}\)). Два зв'язку електронів локалізуються на атомі F, тому ми можемо записати молекулу як Na+ F -. Зауважте, що якби ми збуджували електрон від ψ 1 до ψ 2 за допомогою світла, отримана електронна конфігурація була б (ψ 1 1 ψ 2 1), і ми мали б Na 0 F 0. Це називається переходом передачі заряду.
Короткий зміст молекулярної орбітальної теорії досі:
• Додайте та відніміть хвильові функції AO, щоб зробити MoS. Два АОС → два MoS. У загальному плані загальна кількість МО дорівнює кількості орбіталей на основі АТ.
• Ми показали найпростіший випадок (всього дві базові орбіталі). Більш точні розрахунки використовують набагато більший базисний множина (більше AoS) і вирішують для матриці c, що дає найменшу загальну енергію, використовуючи математично дружні наближення функції потенційної енергії, що є частиною гамільтонового оператора H.
• Більше вузлів → більш висока енергія МО
• Порядок зв'язку = ½ (# зв'язуючих електронів - # антитілірующих електронів)
• Полярність зв'язку виникає на зображенні МО як орбітальний «характер».
• AoS, які знаходяться далеко один від одного в енергії, не сильно взаємодіють, коли вони об'єднуються, щоб зробити МО.