12: Рівноваги розчинності
- Page ID
- 19048

Переконайтеся, що ви добре розумієте наступні основні ідеї:
- Обговорити ролі енергії решітки та гідратації у визначенні розчинності солі у воді.
- Поясніть, що таке якісна схема поділу аналізу, і як вона працює.
- Напишіть вираження розчинності продукту для солі, надавши її формулу.
- Поясніть різницю між іонним продуктом і продуктом розчинності.
- З огляду на формулу солі та її значення K s, обчисліть молярну розчинність.
- Пояснення принципу Ле Шательє призводить до загального іонного ефекту.
- Поясніть, чому сильна кислота, така як HCl, розчиняє помірно розчинну сіль слабкої кислоти, але не сіль сильної кислоти.
- Опишіть, що відбувається (і чому), коли водний аміак повільно додають в розчин нітрату срібла
Розчинення солі у воді - це хімічний процес, який регулюється тими ж законами хімічної рівноваги, які застосовуються до будь-якої іншої реакції. Однак існує ряд особливих аспектів цих рівноваг, які відрізняють їх від більш загальних, які висвітлюються в наборі уроків, присвячених конкретно хімічної рівноваги. До них відносяться такі теми, як загальний іонний ефект, вплив рН на розчинність, перенасичення та деякі особливі характеристики особливо важливих систем розчинності.
Розчинність: розчинення солей у воді
Капніть трохи звичайної кухонної солі в склянку води, і спостерігайте, як вона «зникне». Ми називаємо це розчиненням, і пояснюємо це як процес, при якому одиниці натрію та хлору відриваються від поверхні кристалів, оточуються молекулами H 2 O і стають гідратованими іонами.
\[NaCl_{(s)} \rightarrow Na^+_{(aq)}+ Cl^–_{(aq)} \]
Позначення (aq) означає «водний» і походить від aqua, латинського слова, що позначає вода. Він використовується всякий раз, коли ми хочемо підкреслити, що іони гідратовані - що молекули H 2 O прикріплені до них.
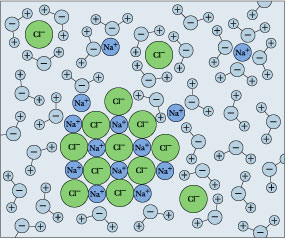
Пам'ятайте, що рівновага розчинності і розрахунки, які стосуються її, мають сенс лише тоді, коли обидві сторони (тверді речовини і розчинені іони) присутні одночасно. Але якщо ви продовжуєте додавати сіль, настане момент, коли вона більше не здається розчиняється. Якщо ця умова зберігається, ми говоримо, що сіль досягла межі розчинності, а розчин насичений в NaCl. Ситуація зараз описується
\[NaCl_{(s)} \rightleftharpoons Na^+_{(aq)}+ Cl^–_{(aq)}\]
в якому тверде тіло і його іони знаходяться в рівновазі.
Сольові розчини, які досягли або перевищили межі розчинності (зазвичай 36-39 г на 100 мл води), відповідають за видатні особливості геохімії землі. Зазвичай вони утворюються при вилуговуванні NaCl з ґрунтів у води, що впадають у солоні озера в посушливих регіонах, які не мають природних виходів; подальше випаровування цих розсолів змушують вищевказану рівновагу вліво, утворюючи природні відкладення солей. Вони часто домішуються з іншими солями, але в деяких випадках є майже чистими NaCl. Багато частин світу містять поховані поклади NaCl (відомий як галіт), які утворилися в результаті випаровування стародавніх морів, і які зараз видобуваються.
Розчинності найбільш принципово виражаються в молярних (моль L -1 розчину) або моляль (моль кг —1 води) одиницях. Але для практичного використання при приготуванні запасних розчинів довідники хімії зазвичай висловлюють розчинності в перерахунку на 100 мл води при заданій температурі, часто відзначаючи останню в верхньому рядку. Таким чином, 6,9 20 означає, що 6,9 г розчиненої речовини розчиняється в 100 мл води при 20° С. При відсутності кількісних даних використовуються позначення «розчинний», «нерозчинний», «слабо розчинний» і «добре розчинний». Немає узгодженого стандарту для цих класифікацій, але корисним орієнтиром може бути те, що показано нижче.
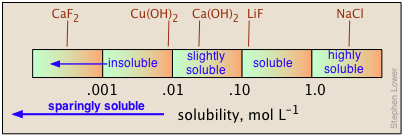
Розчинності солей у воді охоплюють надзвичайно великий діапазон значень, від майже повністю нерозчинних до добре розчинних. Більше того, не існує простого способу прогнозування цих значень або навіть пояснення тенденцій, які спостерігаються для розчинності різних аніонів всередині даної групи періодичної таблиці.

В кінцевому підсумку рушійна сила розчинення (і для всіх хімічних процесів) визначається зміною вільної енергії Гіббса. Але оскільки багато курсів покривають розчинність перед введенням вільної енергії, ми не будемо переслідувати цього тут. Розчинення солі концептуально розуміється як послідовність двох процесів, зображених вище:
- розрив іонної решітки твердого тіла,
- з подальшим приєднанням молекул води до виділених іонів.
Перший крок споживає велику кількість енергії, те, що саме по собі сильно перешкоджало б розчинності. Але другий крок вивільняє велику кількість енергії і, таким чином, має зворотний ефект. Таким чином, чиста зміна енергії залежить від суми двох великих енергетичних термінів (часто наближаються до 1000 кДж/моль), що мають протилежні ознаки. Кожен з цих термінів певною мірою буде впливати розмір, заряд і поляризуваність конкретних іонів, що беруть участь, а також на решітчасту структуру твердого тіла. Така велика кількість змінних унеможливлює прогнозування розчинності даної солі. Тим не менш, існують деякі чіткі тенденції щодо того, як розчинності ряду солей даного аніону (таких як гідроксиди, сульфати тощо) змінюються з групою періодичної таблиці. І звичайно, існує ряд загальних правил розчинності — наприклад, що всі нітрати розчинні, тоді як більшість сульфідів нерозчинні.
Розчинність і температура
Розчинність зазвичай збільшується з температурою - але не завжди. Це дуже очевидно з розчинності проти. -температурні графіки, показані тут. (Деякі сюжети пофарбовані по-різному, щоб було легше розрізнити їх там, де вони скупчуються разом.) Температурна залежність будь-якого процесу залежить від зміни його ентропії — тобто від ступеня, до якого теплова кінетична енергія може поширюватися по всій системі. Коли тверда речовина розчиняється, його складові молекули або іони дифундують в набагато більший обсяг розчину, несучи разом з ними свою теплову енергію. Отже, ми зазвичай очікуємо, що ентропія збільшиться - те, що робить будь-який процес більшою мірою при більш високій температурі.
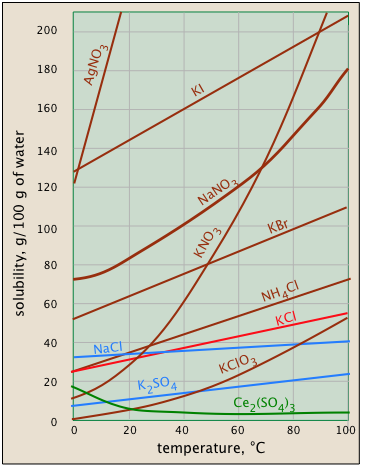
Так чому ж розчинність сульфату церію (зелена ділянка) зменшується з температурою? Розгін самих іонів Ce 3 + і SO 4 2— все ще пов'язаний зі збільшенням ентропії, але в цьому випадку ентропія води зменшується ще більше завдяки впорядкуванню молекул Н 2 О, які приєднуються до Ce 3 + іони, коли вони стають гідратованими. Важко передбачити ці ефекти або пояснити, чому вони виникають в окремих випадках - але вони трапляються.
Важливість помірно розчинних твердих речовин
Всі тверді речовини, які дисоціюються на іони, виявляють певну межу їх розчинності, але ті, чиї насичені розчини перевищують приблизно 0,01 моль L —1, не можуть бути оброблені простими константами рівноваги через утворення іонної пари, що значно ускладнює їх поведінку. З цієї причини більшість з того, що слід в цьому уроці, обмежується солями, які потрапляють в категорію «малорозчинні». Важливість помірно розчинних твердих речовин виникає через те, що утворення такого продукту може ефективно видаляти відповідні іони з розчину, тим самим рухаючи реакцію вправо. Розглянемо, наприклад, що відбувається, коли ми змішуємо розчини селітри стронцію і хлористого калію в мольному співвідношенні 1:2. Хоча ми могли б представляти цей процес
\[Sr(NO_3)_{2(aq)}+ 2 KCl_{(aq)}→ SrCl_{(aq)}+ 2 KNO_{3(aq)} \label{1}\]
чисте іонне рівняння
\[Sr^{2+} + 2 NO_3^– + 2 K^+ + 2 Cl^– → Sr^{2+} + 2 NO_3^– + 2 K^+ + 2 Cl^–\]
вказує на те, що ніяких чистих змін взагалі не відбулося! Звичайно, якби розчин випаровувався до сухості, ми б закінчилися сумішшю чотирьох солей, показаних у Рівнянні\(\ref{1}\), тому в цьому випадку ми можемо сказати, що реакція наполовину повна. Протипоставте це з тим, що станеться, якщо поєднати рівномолярні розчини хлориду барію і сульфату натрію:
\[BaCl_{2(aq)}+ Na_2SO_{4(aq)}→ 2 NaCl_{(aq)}+ BaSO_{4(s)} \label{2}\]
чиє чисте іонне рівняння
\[Ba^{2+} + \cancel{ 2 Cl^–} + \cancel{2 Na^+} + SO_4^{2–} → \cancel{2 Na^+} + \cancel{2 Cl^–} + BaSO_{4(s)}\]
який після скасування, як терміни з обох сторін, стає просто
\[Ba^{2+} + SO_4^{2– }→ BaSO_{4(s)} \label{3}\]
Оскільки утворення помірно розчинних твердих речовин є «повним» (тобто рівноваги, такі як показано вище для сульфату барію, лежать поки що праворуч), практично всі один або обидва сприяють іонам по суті видаляються з розчину. Такі реакції, як кажуть, кількісні, і вони особливо важливі в аналітичній хімії:
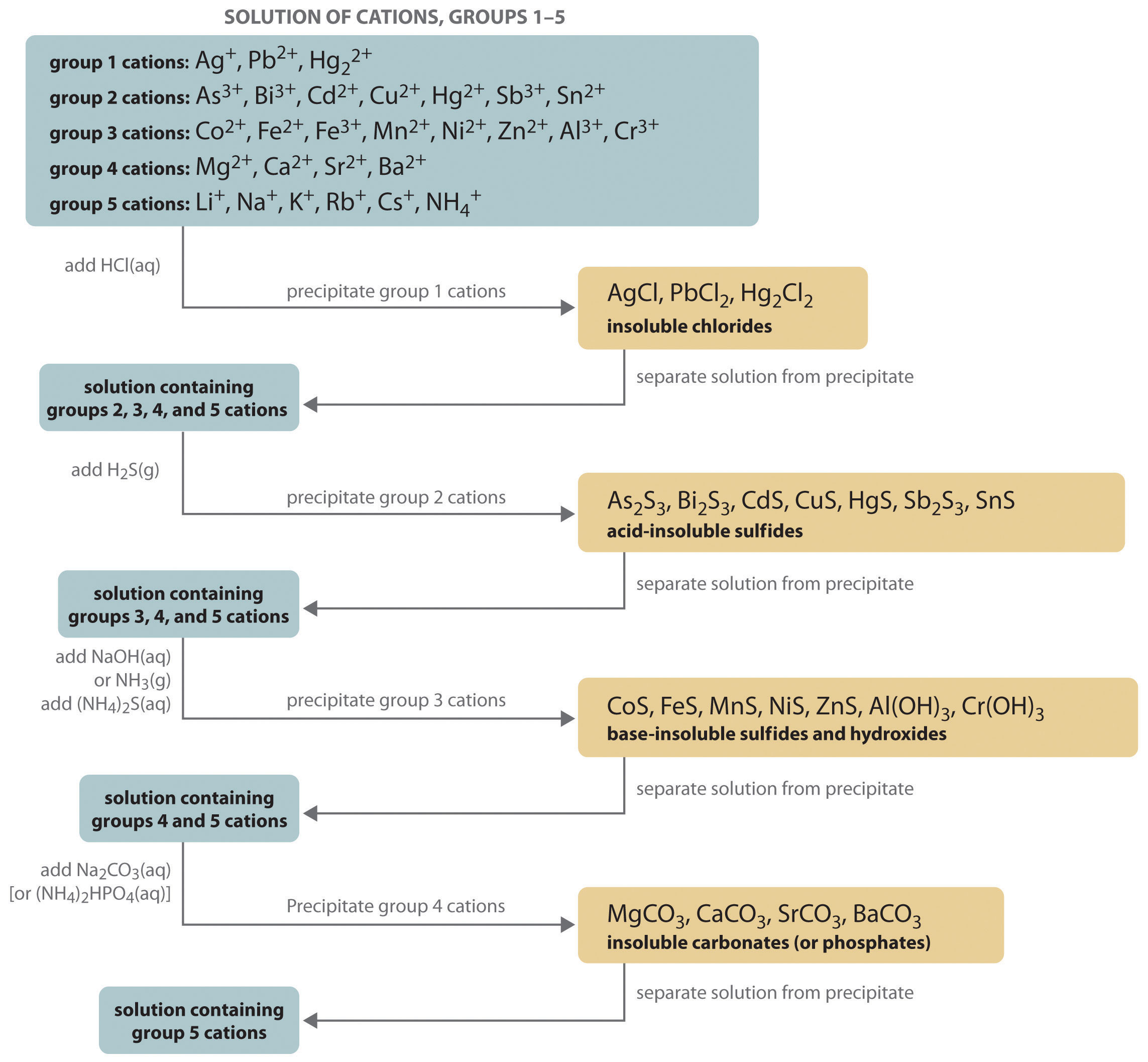
- Якісний аналіз: Це найчастіше відноситься до процедурної схеми, широко зустрічається на перших курсах лабораторних курсів, при якій суміш катіонів (зазвичай у вигляді їх розчинених нітратних солей) систематично відокремлюється і ідентифікується на основі розчинності їх різні аніонні солі, такі як хлориди, карбонати, сульфати та сульфіди. Хоча ця форма якісного аналізу більше не використовується сучасними хіміками (інструментальні методи, такі як атомно-абсорбційна спектроскопія, набагато швидші та всебічні), вона все ще цінується як навчальний інструмент для ознайомлення студентів з деякими основними класами неорганічних солі, і для розвитку основних навичок, пов'язаних із спостереженням, організацією та інтерпретацією результатів у лабораторії.
- Кількісний гравіметричний аналіз: У цій класичній формі хімічного аналізу нерозчинну сіль катіону отримують шляхом осадження її шляхом додавання відповідного аніону. Потім осад збирають, сушать і зважують («гравіметрія») з метою визначення концентрації катіону в зразку. Наприклад, гравіметрична процедура визначення кількості барію у зразку може включати осадження металу як сульфату відповідно до рівняння\(\ref{3}\) вище, використовуючи надлишок сульфатного іона для забезпечення повного видалення барію. Цей метод кількісного аналізу став надзвичайно важливим у другій половині дев'ятнадцятого століття, до якого часу стали доступні досить точні атомні ваги, були розроблені чутливі аналітичні ваги. Це було лише в 1960-х, що він став значною мірою витіснений інструментальними методами, які були набагато швидше і точніше. Гравіметричний аналіз як і раніше зазвичай включається як частина більш просунутих лабораторних інструкцій, багато в чому як засіб розробки ретельної лабораторної техніки.
Продукти розчинності та рівноваги
Деякі солі і подібні сполуки (наприклад, деякі гідроксиди металів) дисоціюють повністю при їх розчиненні, але ступінь їх розчинення настільки обмежена, що отримані розчини проявляють лише дуже слабку провідність. У цих солей, які в іншому випадку діють як сильні електроліти, ми можемо розглядати процес розчинення-дисоціації як справжню рівновагу. Хоча зараз це здається майже тривіальним, це відкриття, зроблене в 1900 році Вальтером Нернстом, який застосував Закон масової дії до схеми дисоціації Арренія, вважається одним з основних кроків у розвитку нашого розуміння іонних рішень.
Використовуючи хромат срібла як приклад, ми виражаємо його розчинення у воді як
\[Ag_2CrO_{4(s)} \rightarrow 2 Ag^+_{(aq)}+ CrO^{2–}_{4(aq)} \label{4a}\]
Коли цей процес досягає рівноваги (що вимагає присутності якогось твердого тіла), ми можемо написати (залишаючи "(aq) s» для простоти)
\[Ag_2CrO_{4(s)} \rightleftharpoons 2 Ag^+ + CrO^{2–}_{4} \label{4b}\]
Константа рівноваги формально
\[K = \dfrac{[Ag^+]^2[CrO_4^{2–}]}{[Ag_2CrO_{4(s)}]} = [Ag^+]^2[CrO_4^{2–}] \label{5a}\]
Але оскільки тверді речовини зазвичай не з'являються у виразах рівноваги, постійна рівноваги для цього процесу є
\[[Ag^+]^2 [CrO_4^{2–}] = K_s = 2.76 \times 10^{–12} \label{5b}\]
Оскільки константи рівноваги такого роду записуються як продукти, отримані К широко відомі як продукти розчинності, позначаються\(K_s\) або\(K_{sp}\).
Власне кажучи, одиниці концентрації не з'являються в рівноважних постійних виразах. Однак багато інструкторів вважають за краще, щоб студенти показували їх у будь-якому випадку, особливо при використанні розчинності продуктів для розрахунку концентрацій. Якщо це зробити,\(K_s\) в Рівнянні\(\ref{5b}\) буде мати одиниці моль 3 L —3.
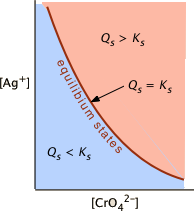
Рівновага і нерівновага в системах розчинності
Такий вираз, як [Ag +] 2 [CrO 4 2—] у відомому загалом як іонний продукт - це іонний продукт для хромату срібла. Іонний продукт в принципі може мати будь-яке позитивне значення, в залежності від концентрацій задіяних іонів. Тільки в особливому випадку, коли його значення ідентичне K s, він стає продуктом розчинності. Рішення, в якому це так, кажуть, насичене. Таким чином, коли
\[[Ag^+]^2 [CrO_4^{2–}] = 2.76 \times 10^{-12}\]
при температурі і тиску, при якому застосовується це\(K_s\) значення, ми говоримо, що «розчин насичений хроматом срібла».
Розчин повинен бути насиченим, щоб бути в рівновазі з твердим тілом. Це необхідна умова рівноваги розчинності, але воно само по собі недостатньо. Справжня хімічна рівновага може виникнути тільки тоді, коли всі компоненти одночасно присутні. Система розчинності може перебувати в рівновазі тільки тоді, коли частина твердого тіла контактує з насиченим розчином його іонів. Нерозуміння цього є дуже частою причиною помилок у вирішенні проблем розчинності.
Ненасичені і перенасичені розчини
Якщо іонний продукт менший за продукт розчинності, система не знаходиться в рівновазі і не може бути присутнім твердого тіла. Такий розчин, як кажуть, є недонасиченим. Пересичений розчин - це той, в якому іонний продукт перевищує продукт розчинності. Пересичений розчин не знаходиться в рівновазі, і жодне тверде тіло зазвичай не може бути присутнім в такому розчині. Якщо додати частину твердої речовини, надлишки іонів випадають в осад і до досягнення рівноваги розчинності.
Як дізнатися стан насичення розчину
Це просто питання порівняння іонного продукту\(Q_s\) з продуктом розчинності\(K_s\). S o для системи
\[Ag_2CrO_{4(s)} \rightleftharpoons 2 Ag^+ + CrO_4^{2–} \label{4ba}\]
рішення, в якому\(Q_s < K_s\) (тобто\(K_s /Q_s > 1\)) є недонасиченим (синє затінення) і не буде присутній твердий. Комбінації [Ag +] і [CrO 4 2—], які відповідають насиченому розчині (і, отже, рівновазі), обмежуються тими, що описані кривою лінією. Рожева область праворуч від цієї кривої представляє перенасичений розчин.
Виявлено, що зразок ґрунтових вод, що просочився через шар гіпсу (CaSO 4, K s = 4.9E—5 = 10 —4,3) має 8,4Е—5 М у Ca 2 + та 7.2E—5 М у SO 4 2—. Яке рівноважний стан даного розчину щодо гіпсу?
Рішення
іонний продукт
\[Q_s = (8.4 \times 10^{–5})(7.2 \times 10^{-5}) = 6.0 \times 10^{–4}\]
перевищує\(K_s\), тому співвідношення K с/Q s > 1 і розчин перенасичений в CaSO 4.
Як визначаються розчинності?
Існує два основних методу, жоден з яких не є настільки надійним для слаборозчинних солей:
- Випарити насичений розчин твердого речовини до сухості, і зважити те, що залишилося.
- Виміряйте електропровідність насиченого розчину, яка буде пропорційна концентрації іонів.
Як розчинність пов'язана з продуктами розчинності
Розчинність (під якою ми зазвичай маємо на увазі молярну розчинність) твердого тіла виражається як концентрація «розчиненого твердого речовини» в насиченому розчині. У випадку простого твердого речовини 1:1, такого як AgCl, це буде просто концентрація Ag + або Cl - в насиченому розчині. Однак для більш складної стехіометрії, такої як хромат срібла, розчинність становила б лише половину концентрації Ag +.
Для прикладу позначимо розчинність Ag 2 CrO 4 як\(S\) моль L —1. Тоді для насиченого розчину ми маємо
- \([Ag^+] = 2S\)
- \( [CrO_4^{2–}] = S\)
Підставивши це в рівняння\(\ref{5b}\) вище,
\[(2S)^2 (S) = 4S^3 = 2.76 \times 10^{–12}\]
\[S= \left( \dfrac{K_s}{4} \right)^{1/3} = (6.9 \times 10^{-13})^{1/3} = 0.88 \times 10^{-4} \label{6a}\]
таким чином розчинність є\(8.8 \times 10^{–5}\; M\).
Зверніть увагу, що співвідношення між розчинністю і постійною розчинності продукту залежить від стехіометрії реакції розчинення. З цієї причини безглуздо порівнювати розчинності двох солей, що мають формули\(A_2B\) і\(AB_2\), виходячи з їх\(K_s\) значень.
Безглуздо порівнювати розчинності двох солей, що мають різні формули на основі їх\(K_s\) значень.
при цих умовах.
Рішення
молі розчиненої речовини в 100 мл; S = 0,0016 г/78,1 г/моль = 2.05E-5 моль
S = 2,05Е—5 моль/0,100 л = 2,05Е-4 М
К с = [Са 2 +] [Ф —] 2 = (С) (2 С) 2 = 4 × (2.05Е—4) 3 = 3.44Е—11
Оцініть розчинність La (IO 3) 3 і розрахуйте концентрацію йодата в рівновазі з твердим йодатом лантану, для якої K s = 6,2 × 10 —12.
Рішення
Рівняння для розчинення дорівнює
\[La(IO_3)_3 \rightleftharpoons La^{3+ }+ 3 IO_3^–\]
Якщо розчинність дорівнює S, то рівноважні концентрації іонів будуть
[La 3 +] = S і [IO 3 —] = 3 S. Тоді К с = [La 3+] [IO 3 —] 3 = S (3 S) 3 = 27 S 4
27 S 4 = 6,2 × 10 —12, S = (((6,2 ÷ 27) × 10 —12) ¼ = 6,92 × 10 —4 М
[ІО 3 —] = 3 S = 2.08 × 10 —5 (М)
Кадмій є високотоксичним забруднювачем навколишнього середовища, який потрапляє в стічні води, пов'язані з виплавкою цинку (Cd і Zn зазвичай зустрічаються разом в рудах ZnS) і в деяких гальванічних процесах. Одним із способів контролю кадмію в потоках стоків є додавання гідроксиду натрію, який осаджує нерозчинний Cd (OH) 2 (K s = 2.5E—14). Якщо 1000 л певної стічної води містить Cd 2+ при концентрації 1,6Е—5 М, яка концентрація Cd 2+ залишиться після додавання 10 л 4 М розчину NaOH?
Рішення
Як і до більшості реальних проблем, до цього найкраще підходити як низка менших проблем, роблячи спрощення наближення відповідно.
Обсяг очищеної води: 1000 л + 10 л = 1010 л
Концентрація ОН — при додаванні до 1000 л чистої води:
(4 М) × (10 Л)/(1010 Л) = 0,040 М
Початкова концентрація Cd 2 + в 1010 л води:
(1,6 Е—5 М) х (100/101) ≈ 1,6 Е—5 М
Найпростіший спосіб вирішити це - почати з припущення, що стехіометрична кількість Cd (OH) 2 утворюється - тобто весь Cd 2 + потрапляє в осад.
| Концентрації | [Кд 2+], М | [О —], М |
|---|---|---|
| ініціал | 1.6Е—5 | 0,04 |
| змінити | —1.6Е—5 | —3.2Е—5 |
| фінал: | 0 | 0.04 — 3.2Е—5 ≈ .04 |
Тепер «увімкніть рівновагу» — знайдіть концентрацію Cd 2 +, яка може існувати в 0,04 М ОН — розчині:
| Концентрації | [Кд 2+], М | [О —], М |
|---|---|---|
| ініціал | o | 0,04 |
| змінити | + х | +2 х |
| при рівновазі | х | 0,04 + 2 х ≈ .04 |
Замініть ці значення у вираз розчинності продукту:
\[Cd(OH)_{2(s) } = [Cd^{2+}] [OH^–]^2 = 2.5 \times 10^{–14}\]
\[[Cd^{2+}] = \dfrac{2.5 \times 10^{–14}}{ 16 \times 10^{–4}} = 1.6 \times 10^{–13}\; M\]
Відзначимо, що стоки тепер будуть дуже лужними:
\[pH = 14 + \log 0.04 = 12.6\]
тому для того, щоб відповідати екологічним нормам, необхідно додати еквівалентну кількість сильної кислоти для нейтралізації води перед її випуском.
Все лише спрощення реальності
Прості відносини між K s та молярною розчинністю, викладені вище, та приклади розрахунку, наведені тут, не можна покладатися, щоб дати правильні відповіді. Деякі з причин цього пояснюються в частині 2 цього уроку, і в основному пов'язані з неповною дисоціацією багатьох солей і з комплексним утворенням у присутності аніонів, таких як Cl - і OH -. Ситуація добре описана в статті Що ми повинні навчити початківців про продукти розчинності та розчинності? Стівен Хоукс (J Chem Educ. 1998 75 (9) 1179-81). Дивіться також попередню статтю Meites, Pode та Thomas Чи пов'язані продукти розчинності та розчинності? (J Хім Едук. 1966 43 (12) 667-72).
Виявляється, рівноваги розчинності частіше, ніж не включають багато конкуруючих процесів, і їх суворе лікування може бути досить складним. Тим не менш, важливо, щоб студенти освоїли ці надто спрощені приклади. Однак важливо і те, щоб їх не сприймали занадто серйозно!
Загальний іонний ефект
Давно відомо, що розчинність помірно розчинної іонної речовини помітно знижується в розчині іншого іонного з'єднання, коли дві речовини мають загальний іон. Це саме те, що можна було б очікувати на основі принципу Ле Шательє; всякий раз, коли процес
\[CaF_{2(s)} \rightleftharpoons Ca^{2+} + 2 F^– \label{7}\]
знаходиться в рівновазі, додавання більшої кількості іона фтору (у вигляді добре розчинних NaF) змістить склад вліво, зменшуючи концентрацію Са 2 +, і тим самим ефективно зменшуючи розчинність твердого речовини. Ми можемо виразити це кількісно, зазначивши, що вираження розчинності продукту
\[[Ca^{2+}][F^–]^2 = 1.7 \times 10^{–10} \label{8}\]
повинен завжди триматися, навіть якщо деякі з іонних видів походять з інших джерел, ніж CaF 2 (s). Наприклад, якщо до розчину спочатку в рівновазі з твердим CaF 2 додається деяка кількість x іона фтору, ми маємо
- \([Ca^{2+}] = S\)
- \([F^–] = 2S + x\)
так що
\[K_s = [Ca^{2+}][ F^–]^2 = S (2S + x)^2 . \label{9a}\]
\[K_s ≈ S x^2 \]
або
\[S ≈ \dfrac{K_s}{x^2} \label{9b}\]
Студенти університетського рівня повинні мати можливість вивести ці відносини для іонних твердих тіл будь-якої стехіометрії.
Наведені нижче графіки ілюструють загальний іонний ефект для хромату срібла, оскільки концентрація іонів хромату збільшується додаванням розчинного хромату, такого як\(Na_2CrO_4\).
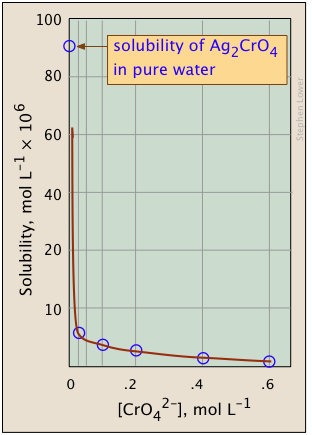
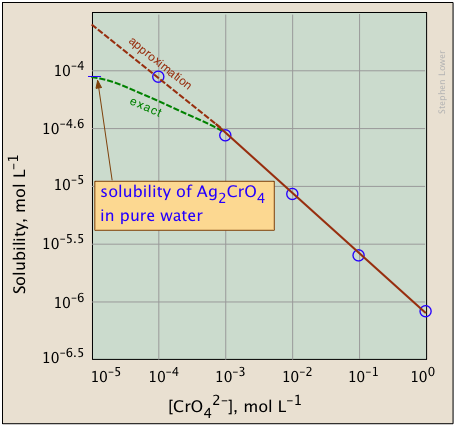
Чим відрізняється сюжет справа? Якщо уважно подивитися на шкали, то побачите, що ця побудована логарифмічно (тобто в степенях 10.) Зверніть увагу, як набагато ширший діапазон значень може відображатися на логарифмічному графіку. Сенс показу цієї пари графіків полягає в тому, щоб проілюструвати велику корисність графіків концентрації журналу в рівноважних розрахунках, в яких прості наближення (наприклад, зроблені в Рівнянні,\(\ref{9b}\) можуть давати прямі лінії в діапазоні значень, для яких наближення є дійсним.
Обчисліть розчинність сульфату стронцію (K s = 2,8 × 10 -7) в
- чиста вода і
- в 0,10 моль л -1 розчин\(Na_2SO_4\).
\[S = \sqrt{K_s} = \sqrt{ 2.8 \times 10^{–7} } = 5.3 \times 10^{–4}\]
(б) У 0,10 моль L —1 Na 2 SO 4 ми маємо
= [Sr 2 +] [СО 4 2—] = S × (0,10 + S) = 2,8 × 10 —7Оскільки S є незначним порівняно з 0,10 М, ми робимо наближення
= [Sr 2 +] [СО 4 2—] ≈ S × (0,10 М) = 2,8 × 10 —7тому
Це приблизно в 100 разів менше, ніж результат з (а).
Вибіркові опади та поділи
Відмінності в розчинності широко використовуються для вибіркового видалення одного виду з розчину, що містить кілька видів іонів.
Продукти розчинності AgCl і Ag 2 CrO 4 становлять 1,8Е—10 і 2,0Е—12 відповідно. Припустимо, що розведений розчин AgNO 3 додають крапельно до розчину, що містить 0,001 M Cl — і 0,01 M CrO 4 2—.
- Яке тверде речовина, AgCl або Ag 2 CrO 4, випаде в осад першим?
- Яка фракція першого аніону буде видалена, коли друга тільки почне осаджуватися? Нехтувати будь-якими змінами гучності.
Рішення
Концентрації іонів срібла, необхідні для осаду двох солей, виявляються шляхом заміни у відповідні вирази продукту розчинності:
- випасти в осад AgCl: [Аг +] = 1,8Е-10/0,001 = 1,8Е-7 М
- випасти в осад Ag 2 CrO 4: [Аг +] = (2,0Е-12/ 0,01) ½ = 1,4Е—5 М
Першим твердим речовиною, що утворюється при збільшенні концентрації Ag +, буде AgCl. З часом концентрація Ag + досягає 1.4E-5 M і Ag 2 CrO 4 починає осаджуватися. У цей момент концентрація іона хлориду в розчині становитиме 1,3Е-5 М, що становить близько 13% від кількості, спочатку присутньої.
Попередній приклад є основою титрування хлориду Мора Ag +, зазвичай робиться для визначення солоності проб води. Точка еквівалентності цього титрування опадів виникає, коли більше не утворюється AgCl, але немає можливості спостерігати це безпосередньо за наявності білого AgCl, який підвішений у контейнері. Перед початком титрування в розчин додають невелику кількість К 2 CrO 4. Ag 2 CrO 4 має червоно-оранжевий колір, тому його утворення, яке сигналізує про приблизне закінчення опадів AgCl, можна виявити візуально.
Конкуруючі рівноваги за участю твердих
Вираз розчинності, ймовірно, є винятком, а не правилом. Такі рівноваги часто конкурують з іншими реакціями з такими видами, як H + або OH -, комплексоутворювачі, окислення-відновлення, утворення інших слаборозчинних видів або, у випадку карбонатів і сульфітів, газоподібних продуктів. Точні обробки цих систем можуть бути надзвичайно складними, передбачаючи розв'язування великих множин одночасних рівнянь. Для більшості практичних цілей досить визнати загальні тенденції, і провести приблизні розрахунки.
Солі слабких кислот розчинні в сильних кислотах, але сильні кислоти не будуть розчиняти солі сильних кислот
Розчинність легкорозчинної солі слабкої кислоти або основи буде залежати від рН розчину. Щоб зрозуміти причину цього, розглянемо гіпотетичну сіль МА, яка розчиняється, утворюючи катіон M +, і аніон А - який також є кон'югатною основою слабкої кислоти HA. Той факт, що кислота слабка, означає, що іони водню (завжди присутні у водних розчині) і катіони M + будуть конкурувати за A -:

Чим слабкіше кислота ГК, тим охочіше буде відбуватися реакція, тим самим з'їдаючи А-іони. Якщо надлишок Н + стає доступним шляхом додавання сильної кислоти, ще більше А - іонів буде витрачено, врешті-решт зворотну реакцію
відбуватися реакція, тим самим з'їдаючи А-іони. Якщо надлишок Н + стає доступним шляхом додавання сильної кислоти, ще більше А - іонів буде витрачено, врешті-решт зворотну реакцію , змушуючи тверду речовину розчинятися.
, змушуючи тверду речовину розчинятися.

У, наприклад, сульфат-іони вступають в реакцію з іонами кальцію з утворенням нерозчинних CaSO 4. Додавання сильної кислоти, такої як HCl (яка повністю дисоційована) не має ефекту, оскільки CaCl 2 розчинний. Хоча H + може протонувати деякі іони SO 4 2— з утворенням сульфату водню («бісульфату») HSO 4 -, ця амфолітна кислота занадто слабка, щоб повернути назад, витягуючи значну частку сульфатних іонів з CaSO 4 (s).
Обчисліть концентрацію іона алюмінію в розчині, який знаходиться в рівновазі з гідроксидом алюмінію, коли рН тримається на рівні 6,0.
Рівноваги є
\[Al(OH)_3 \rightleftharpoons Al^{3+} + 3 OH^–\]
із
\[K_s = 1.4 \times 10^{–34}\]
і
\[H_2O \rightleftharpoons H^+ + OH^–\]
із
\[K_w = 1 \times 10^{–14}\]
Підставивши вираз рівноваги для другого з них на те, що для першого, отримаємо
\[[OH^–]^3 = \left( \dfrac{K_w}{ [H^+]}\right)^3 = \dfrac{K_s}{[Al^{3+}]}\]
(1,0 × 10 —14)/(1,0 × 10 —6) 3 = (1,4 × 10 —24)/[Аль 3 +]
з якого ми знаходимо
\[[Al^{3+}] = 1.4 \times 10^{–10}\; M\]
Формування конкуруючого осаду
Якщо два різних аніони конкурують з одним катіоном, утворюючи два можливі опади, результат залежить не тільки від розчинності двох твердих тіл, але і від концентрацій відповідних іонів.
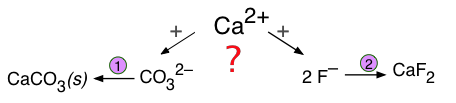
Такі змагання особливо важливі в грунтових водах, які набувають розчинені речовини з різних джерел, коли вони проходять через шари осаду, що мають різний склад. Як показує наступний приклад, конкуруючі рівноваги цих видів дуже важливі для розуміння геохімічних процесів, пов'язаних з утворенням і перетворенням родовищ корисних копалин.
Припустимо, що грунтові води, що містять 0,001 М F — і 0,0018 М СО 3 2— просочуються через осад, що містить кальцит, СаСО 3. Чи буде кальцит замінений флюоритом, CaF 2?
Два рівноваги розчинності
\[\ce{CaCO3 <=> Ca^{2+} + CO3^{2–} \quad K_s = 10^{–8.1}\]
\[\ce{CaF2 <=> Ca^{2+} + 2 F^{–} \quad K_s = 10^{–10.4}\]
Рішення:
Рівновага між двома твердими речовинами та двома аніонами є
\[CaCO_3 + 2 F^–\rightleftharpoons CaF_2 + CO_3^{2–}\]
Це всього лише сума реакції розчинення для CaCo 3 і зворотна для CaF 2, тому постійна рівноваги дорівнює
\[K = \dfrac{[CO_3^{2–}]}{ [F^–]^2} = \dfrac{10^{–8.1}}{ 10^{–10.4}} = 200\]
Тобто, два твердих тіла можуть співіснувати тільки в тому випадку, якщо коефіцієнт реакції Q ≤ 200. Підставляючи задані концентрації іонів, ми виявляємо, що
\[Q = \dfrac{0.0018}{0.0012} = 1800\]
Починаючи з Q > K, можна зробити висновок, що кальцит не зміниться на флюорит.
Комплексне утворення іонів
Більшість іонів перехідних металів мають порожні d орбіталі, які мають достатньо низьку енергію, щоб мати можливість приймати електронні пари від донорів електронів від катіонів, в результаті чого утворюється ковалентно зв'язаний комплексний іон. Навіть нейтральні види, які мають незв'язну електронну пару, можуть зв'язуватися з іонами таким чином. Вода є активним донором електронів такого роду, тому водні розчини іонів, таких як Fe 3 + (aq) і Cu 2 + (aq) існують як октаедричні комплекси Fe (H 2 O) 6 3+ і Cu (Н 6 О) 6 2+ відповідно.
Багато зауважень, зроблених вище щодо співвідношення між K s та розчинністю, також стосуються розрахунків, пов'язаних із складним утворенням. Див. Стаття Стівена Хокса Розрахунки комплексоутворення гірші, ніж марні («... до абсурду... і не слід вчити» на вступних курсах.) (J Хім Едук. 1999 76 (8) 1099-1100). Однак дуже важливо, щоб ви розуміли принципи, викладені в цьому розділі.
H 2 O є лише одним можливим донором електронів; NH 3, CN - і багато інших видів (спільно відомих як ліганди) мають самотні пари, які можуть займати вакантні d орбіталі на металевому іоні. Багато з них набагато міцніше зв'язуються з металом, ніж H 2 O, який зазнає зміщення і заміщення одним або декількома з цих лігандів, якщо вони присутні в досить високій концентрації.
Якщо поміщати в контакт з розчином, що містить ліганд, який може зв'язуватися з іоном металу набагато сильніше, ніж Н 2 О, тоді буде сприяти утворенню комплексного іона і розчинність твердого тіла буде більшою. Можливо, найбільш часто зустрічається приклад цього відбувається, коли аміак додають в розчин нітрату міді (II), в якому іон Cu 2 + (aq) сам по собі є комплексним іоном гексаако комплексу, показаним зліва:

Оскільки аміак є слабкою основою, перше, що ми спостерігаємо - це утворення каламутного осаду Cu (ОН) 2 в синьому розчині. Оскільки додається більше аміаку, цей осад розчиняється, і розчин стає інтенсивним глибоким синім кольором, який є кольором гексаминомеду (II) та різних інших споріднених видів, таких як Cu (H 2 O) 5 (NH 3) 2+, Cu (H 2 O) 4 (NH 3) 2 2+ і т.д.

У багатьох випадках комплексообразователя і аніон малорозчинної солі ідентичні. Це особливо схильне траплятися з нерозчинними хлоридами, і це означає, що додавання хлориду для осаду металевого іона, такого як Ag +, спочатку призведе до осаду, але після додавання надлишку Cl - осад буде повторно розчинятися, коли утворюються складні іони.
Деякі важливі системи розчинності
У цьому розділі ми обговорюємо рівноваги розчинності, які стосуються деяких дуже часто зустрічаються аніонів металевих солей. Це особливо стосується видів розлуки, які більшість студентів коледжів зобов'язані проводити (і розуміти!) на перших курсах лабораторних курсів.
Розчинність оксидів і гідроксидів
Оксиди металів і гідроксиди утворюють розчини, що містять ОН — іони. Наприклад, розчинності [слабо розчинного] оксиду і гідроксиду магнію представлені
\[Mg(OH)_{2(s)} → Mg^{2+} + 2 OH^– \label{10}\]
\[MgO_{(S)} + H_2O → Mg^{2+} + 2 OH^– \label{11}\]
Якщо виписати вирази продукту розчинності для цих двох реакцій, ви побачите, що вони ідентичні за формою і значенням.
Нагадаємо, що рН = —лог 10 [Н +], так що [Н +] = 10 —рН.
Можна наївно очікувати, що розчинення оксиду, такого як MgO, дасть в якості одного з його продуктів оксид-іон O 2+. Але іон оксиду є настільки міцною основою, що він захоплює протон з води, утворюючи замість цього два гідроксидних іона:
\[O^{2+} + H_2O → 2 OH^–\]
Це приклад правила, що гідроксид-іон є найсильнішою основою, яка може існувати у водному розчині. 2 «- рівноважна суміш гідратованих молекул СО 2 і вугільної кислоти. Щоб речі були максимально простими, ми не будемо розрізняти їх у наступному, а просто використаємо формулу H 2 CO 3 для колективного представлення двох видів.
Інші метали групи 2, особливо Mg, разом із залізом та декількома іншими перехідними елементами також знаходяться в карбонатних відкладах. Коли дощ падає через повітря, він поглинає атмосферний вуглекислий газ, невелика частина якого вступає в реакцію з водою з утворенням вугільної кислоти. При цьому вся чиста вода, що контактує з повітрям, стає кислою, з часом досягаючи рН 5,6.
Як зазначалося вище, рівновага між бікарбонатними і карбонатними іонами залежить від рН. Оскільки шкала рН є логарифмічною, має сенс (і значно спрощує побудову ділянки) використовувати шкалу журналу для концентрацій. Графік, показаний нижче, відповідає загальній концентрації карбонатної системи 10 —3 М, що є репрезентативним для багатьох грунтових вод. Для річкових та озерних вод 10-5 М було б більш типовим; це просто змістило б криві вниз, не впливаючи на їх форми.

Точки 1 і 2, де суміжні криві перекриваються, відповідають двом pK. Нагадаємо, що коли рН такий же, як pK, концентрації двох кон'югатних видів ідентичні і половина загальної концентрації системи. Це ставить точки кросовера на log 0.5 = —0.3 нижче рівня концентрації системи.
10-3 М розчин бікарбонату натрію мав би рН, позначений точкою 3, причому [H 2 CO 3] і [CO 3 2—] складають лише 1% (10 —5 М) системи. Це відповідає рівновазі
\[2 HCO_3^– \rightleftharpoons H_2CO_3 + CO_3^{2–}\]
Carbonates act as bases and, as such, react with acids. Thus, the portion of the global water cycle that transports carbon from the air into natural waters constitutes a gigantic acid-base reaction that yields hydrogen carbonate ions, commonly referred to as bicarbonate. The natural waters that result have pH values between 6 and 10 and are essentially solutions of bicarbonates.
Limestone caves and sinkholes
When rainwater permeates into the soil, it can become even more acidic owing to the additional CO2 produced by soil organisms. Also, the deeper the water penetrates, the greater its hydrostatic pressure and the more CO2 it can hold, further increasing its acidity. If this water then works its way down through the fissures and cracks within a limestone layer, it will dissolve some of limestone, leaving void spaces which may eventually grow into limestone caves or form sinkholes that can swallow up cars or houses.
A well-known feature of limestone caves is the precipitated carbonate formations that decorate the ceilings and floors. These are known as stalactites and stalagmites, respectively. When water emerges from the ceiling of a cave that is open to the atmosphere, some of the excess CO2 it contains is released as it equilibrates with the air. This raises its pH and thus reduces the solubility of of the carbonates, which precipitate as stalactites. Some of the water remains supersaturated and does not precipitate until it drips to the cave floor, where it builds up the stalagmite formations.
Hard Water
This term refers to waters that, through contact with rocks and sediments in lakes, streams, and especially in soils (groundwaters), have acquired metallic cations such as Ca2+, Mg2+, Fe2+, Fe3+, Zn2+ Mn2+, etc. Owing to the ubiquity of carbonate sediments, the compensating negative charge is frequently supplied by the bicarbonate ion HCO3–, but other anions such as SO42–, F–, Cl–, PO43– and SiO42– may also be significant.
Solid bicarbonates are formed only by Group 1 cations and all are readily soluble in water. But because HCO3– is amphiprotic, it can react with itself to yield carbonate:
\[2 HCO_3^– → H_2O + CO_3^[2–} + CO_{2(g)}\]
If bicarbonate-containing water is boiled, the CO2 is driven off, and the equilibrium shifts to the right, causing any Ca2+ or similar ions to form a cloudy precipitate. If this succeeds in removing the "hardness cations", the water has been "softened". Such water is said to possess carbonate hardness, sometimes known as "temporary hardness". Waters in which anions other than HCO3– predominate cannot be softened by boiling, and thus possess non-carbonate hardness or "permanent hardness".
Hard waters present several kinds of problems, both in domestic and industrial settings:
- Waters containing dissolved salts leave solid deposits when they evaporate. Residents of areas having hard water (about 85 percent of the U.S.) notice evaporative deposits on shower walls, in teakettles, and on newly-washed windows, glassware, and vehicles.
- Much more seriously from an economic standpoint, evaporation of water in boilers used for the production of industrial steam leaves coatings on the heat exchanger surfaces that impede the transfer of heat from the combustion chamber, reducing the thermal transfer efficiency. The resultant overheating of these surfaces can lead to their rupture, and in the case of high-pressure boilers, to disastrous explosions. In the case of calcium and magnesium carbonates, the process is exacerbated by the reduced solubility of these salts at high temperatures. Removal of boiler scales is difficult and expensive.
- Municipal water supplies in hard-water areas tend to be supersaturated in hardness ions. As this water flows through distribution pipes and the plumbing of buildings, these ions often tend to precipitate out on their interior surfaces. Eventually, this scale layer can become thick enough to restrict or even block the flow of water through the pipes. When scale deposits within appliances such as dishwashers and washing machines, it can severely degrade their performance.
- Cations of Group 2 and above react with soaps, which are sodium salts of fatty acids such as stearic acid, C17H35COOH. The sodium salts of such acids are soluble in water, which allows them to dissociate and act as surfactants:
\[C_{17}H_{35}COONa → C_{17}H_{35}COO^– Na^+\]
but the presence of polyvalent ions causes them to form precipitates
\[2 C_{17}H_{35}COO^– + Ca^{2+} → (C_{17}H_{35}COO^–)_2Ca_{(s)}\]
Calcium stearate is less dense than water, so it forms a scum that floats on top of the water surface; anyone who lives in a hard-water area is likely familiar with the unsightly "bathtub rings" it leaves around the high-water mark or the shower-wall stains.
Solubility Complications
All heterogeneous equilibria, on close examination, are beset with complications. But solubility equilibria are somewhat special in that there are more of them. Back in the days when the principal reason for teaching about solubility equilibria was to prepare chemists to separate ions in quantitative analysis procedures, these problems could be mostly ignored. But now that the chemistry of the environment has grown in importance — especially that relating to the ocean and natural waters — there is more reason for chemical scientists to at least know about the limitations of simple solubility products. This section will offer a quick survey of the most important of these complications, while leaving their detailed treatment to more advanced courses.
Tabulated Ks values are notoriously unreliable
Many of the \(K_s\) values found in tables were determined prior to 1940 (some go back to the 1880s!) at a time before highly accurate methods became available. Especially suspect are many of those for highly insoluble salts which are more difficult to measure. A table showing the variations in \(K_{sp}\) values for the same salts among ten textbooks was published by Clark and Bonikamp in J Chem Educ. 1998 75(9) 1183-85.A good An example that used a variety of modern techniques to measure the solubility of silver chromate was published by A.L. Jones et al in the Australian J. of Chemistry, 1971 24 2005-12.
Generations of chemistry students have amused themselves by comparing the disparate Ks values to be found in various textbooks and table. In some cases, they differ by orders of magnitude. There are several reasons for this in addition to the ones described in detail further on.
- The most direct methods of measuring solubilities tend to not be very accurate for sparingly soluble salts. Two-significant figure precision is about the best one can hope in a single measurement.
- Many insoluble salts can exist in more than one crystalline form (polymorphs), and in some cases also as amorphous solids. Precipitation under different conditions (in the presence of different ions, at different temperatures, etc.) can yield different or mixed polymorphs.
- Other ions present in the solution can often get incorporated into the crystalline solid, usually replacing an ion of similar size (substitutional solid solutions). When this happens, it is no longer valid to write the equilibrium condition as a simple "product". This is very common in mineral deposits, and an important consideration in geochemistry,
Most salts are not Completely Dissociated in Water
The dissolution of cadmium iodide is water is commonly represented as
\[CdI_{2(s)} → Cd^{2+} + 2 I^–\]
Firstly, they combine to form neutral, largely-covalent molecular species:
\[Cd^{2+}_{(aq)} + 2 I^–_{(aq)} → CdI_{2(aq)}\]
This non-ionic form accounts for 78% of the Cd present in the solution! In addition, they form a molecular ion \(CdI^–_{(aq)}\) according to the following scheme:
| \(CdI_{2(s)} \rightleftharpoons Cd^{2+} + 2 I^–\) | \(K_1 = 10^{–3.9}\) |
| \(Cd^{2+} + I^– \rightleftharpoons CdI^+\) | \(K_2= 10^{+2.3}\) |
| \(CdI2_{(s)} \rightleftharpoons CdI^++ I^–\) | \(K = 10^{–1.6} = 0.023\) |
The data shown Tables \(\PageIndex{1}\) and \(\PageIndex{2}\) are taken from the article Salts are Mostly NOT Ionized by Stephen Hawkes: 1996 J Chem Educ. 73(5) 421-423. This fact was stated by Arrhenius in 1887, but has been largely ignored and is rarely mentioned in standard textbooks.
As a consequence, the concentration of "free" Cd2+(aq) in an aqueous cadmium iodide solution is only about 2% of the value you would calculate by taking K1 as the solubility product. The principal component of such as solution is actually [covalently-bound] CdI2(aq). It turns out that many salts, especially those of metals beyond Group 2, are similarly only partially ionized in aqueous solution:
| salt | molarity | % cation | other species |
|---|---|---|---|
| KCl | 0.52 | 95 | KCl(aq) 5% |
| MgSO4 | 0.04 | 58 | MgSO4(aq) 42% |
| CaCl2 | 0.44 | 70 | CaCl+(aq) 30% |
| CuSO4 | 0.045 | 56 | CuSO4(aq) 44% |
| CdI2 | 0.50 | 2 | CdI2(aq) 76%, CdI–(aq) 22% |
| FeCl3 | 0.1 | 10 | FeCl2+(aq) 42%, FeCl2(aq) 40%, FeOH2+(aq) 6%, Fe(OH)2+(aq) 2% |
If you are enrolled in an introductory course and do not plan on taking more advanced courses in chemistry or biochemistry, you can probably be safe in ignoring this, since your instructor and textbook likely do so. However, if you expect to do more advanced work or teach, you really should take note of these points, since few textbooks mention them.
Formation of Hydrous Complexes
Transition metal ions form a large variety of complexes with H2O and OH–, both of which have electron-pairs available to coordinate with the central ion. This gives rise to a large variety of soluble species that are in competition with an insoluble solid. Because of this, a single equilibrium constant (solubility product) cannot describe the behavior of a solid such as Fe(OH)3, which we summarize here as an example.
Aquo complexes: The electrostatic field of the positively-charged metal ion enhances the acidic nature of these H2O molecules, encouraging them to shed a proton and leaving OH– groups in their place.
\[Fe(H_2O)_6^{3+} → Fe(H_2O)_5(OH)^{2+}+H^+\]
This is just the first of a series of similar reactions, each one having a successively smaller equilibrium constant:
\[Fe(H_2O)_5(OH)^{2+}→ Fe(H_2O)_4(OH)_2^+→ Fe(H_2O)_3(OH)_3 → Fe(H_2O)_2(OH)_4^-\]
Hydroxo complexes: But there's more: when the hydroxide ion acts as a ligand, it gives rise to a series of hydroxo complexes, of which the insoluble Fe(OH)3 can be considered a member:
| Fe3+ + 3 OH– → Fe(OH)3(s) | 1/Ks = 1038 |
| Fe3+ + H2O → Fe(OH)2+ + H+ | K = 10–2.2 |
| Fe3+ + 2H2O → FeOH+ + 2H+ | K = 10–6.7 |
| Fe3+ + 4H2O → Fe(OH)4– + 4H+ | K = 10–23 |
| 2Fe3+ + 2H2O → Fe2(OH)24+ + 2H+ | K = 10–2.8 |

Перелічені вище рівноваги включають H + і OH - іони, і тому залежать від рН, як показано прямими лініями на графіку, нахили яких відображають залежність рН відповідних іонних видів. При будь-якому заданому рН рівновага з твердим Fe (OH) 3 контролюється іонними видами, що мають найвищу концентрацію при будь-якому заданому рН. Таким чином, відповідні лінії на графіку окреслюють область (позначену помаранчевим затіненням), в якій може існувати тверде тіло.
Іонні взаємодії: «Непоширений іонний ефект»
Легкорозчинна сіль буде більш розчинна в розчині, який містить іони, що не беруть участь. Це якраз протилежність загальному іонному ефекту, і спочатку це може здатися досить неінтуїтивним: чому додавання більшої кількості іонів будь-якого роду змушує сіль швидше розчинятися?
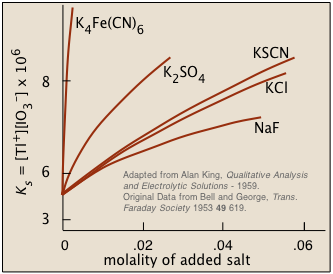
Малюнок\(\PageIndex{2}\) : Розчинність йодата талію в розчині, що містять розчинені солі
Підказку до відповіді можна знайти в іншому факті: чим вище заряд чужорідного іона, тим більш виражений ефект. Це говорить нам про те, що міжіонні (і, отже, електростатичні) взаємодії повинні відігравати певну роль. Деталі досить складні, але загальна ідея полягає в тому, що всі іони в розчині, крім того, що володіють щільно утримуваними водами гідратації, мають тенденцію притягувати навколо себе протилежно заряджені іони («контріони»). Ця «атмосфера» протиіонів завжди досить дифузна, але набагато менше (і більш щільно пов'язана), коли один або обидва види іонів мають більші заряди. Здалеку ці іонно-протиіонні тіла здаються майже електрично нейтральними, що утримує їх від взаємодії між собою (як утворюється осад).

Загальний ефект полягає в зменшенні концентрацій менш екранованих іонів, які доступні для об'єднання, утворюючи осад. Ми говоримо, що термодинамічно-ефективні концентрації цих іонів менші, ніж їх «аналітичні» концентрації. Хіміки називають ці ефективні концентрації іонними активностями, і вони позначають їх фігурними дужками {Ag +} на відміну від квадратних дужок [Ag +], які стосуються номінальних або аналітичних концентрацій.
Хоча концентрації іонів в рівновазі з легкорозчинним твердим тілом настільки низькі, що вони по суті такі ж, як і діяльність, наявність інших іонів при концентраціях близько 0,001M або більше може суттєво зменшити діяльність продуктів розчинення, дозволяючи розчинності повинні бути більшими, ніж передбачали б прості розрахунки рівноваги.
Вимірювані розчинності середні і залежать від розміру
Гетерогенний характер реакцій розчинення призводить до ряду своєрідних ефектів, що стосуються природи рівноваг за участю поверхонь. Вони виникають через те, що схильність кристалічної твердої речовини до розчинення буде залежати від конкретної грані або місця, з якого відбувається розчинення. Оскільки всі кристали представляють різні грані до рішення, виміряний K s дійсно є середнім значенням для цих різних граней.
А оскільки багато солей можуть проявляти різну зовнішню форму в залежності від умов, в яких вони утворюються, продукти розчинності аналогічно залежать від цих умов.
Дуже дрібні кристали більш розчинні, ніж великі
Молекули або іони, розташовані на краях або кутах, менш сильно пов'язані з рештою твердого тіла, ніж ті, що знаходяться на плоских поверхнях, і, отже, мають тенденцію розчинятися легше. Таким чином, крайня ліва грань у схематичній решітці нижче матиме більше граничних молекулярних одиниць, ніж дві інші, і ця грань (11) буде більш розчинною.
Це означає, серед іншого, що менші кристали, у яких співвідношення країв і кутів більше, як правило, матимуть більші значення K s, ніж більші. Як наслідок, більш дрібні кристали, як правило, зникнуть на користь більших. Практичне застосування іноді застосовується, коли осад, спочатку утворився при хімічному аналізі або поділі, занадто дрібний, щоб його можна було видалити фільтрацією. Суспензія витримується при високій температурі протягом декількох годин, з часом кристаліти збільшуються в розмірах. Цю процедуру іноді називають травленням.
Формування пересичених розчинів
Всупереч тому, що вас, можливо, навчили, опади не утворюються, коли продукт концентрації іонів досягає продукту розчинності солі в чистому і спочатку ненасиченому розчині; для утворення осаду з однорідного розчину потрібна певна ступінь перенасичення. Ступінь перенасичення, необхідна для ініціювання опадів, може бути напрочуд великою. При цьому утворення сульфату барію BasO 4 шляхом об'єднання двох видів іонів не відбувається до тих пір, поки Q s не перевищить K s в 160 і більше разів. Частково це відображає той факт, що осадження протікає низкою реакцій, що починаються з утворення іонної пари, яка з часом стає іонним скупченням:
Ба 2 + СО 4 2— → (БаСО 4) 0 → (БаСО 4) 2 0 → (БаСО 4) 3 0 → і т.д.
Завдяки загальній нейтральності ці агрегати не стабілізуються гідратацією, тому вони швидше розпадаються, ніж ні. Але деякі з них можуть врешті-решт вижити, поки вони не стануть досить великими (але все ще субмікроскопічними за розміром), щоб служити ядрами опадів.
Багато речовин, крім солей, утворюють перенасичені розчини, а деякі солі утворюють їх легше, ніж інші. Пересичені розчини легко виготовляються шляхом розчинення твердої речовини до майже межі його розчинності в підігрітому розчиннику, а потім даючи йому охолонути.
K s) і за своєю суттю нестабільні; скидання «насіннєвого» кристала твердого тіла в такий розчин зазвичай ініціює швидке осадження. Але як пояснюється нижче, навіть крихітної частинки пилу може бути достатньо. Стара аптечна хитрість полягає в тому, щоб використовувати кінчик скляного перемішувального стрижня, щоб зішкребти внутрішню поверхню контейнера, що тримає перенасичений розчин; дрібні частинки скла, які виділяються імовірно, служать ядрами опадів.
Проблема зародження: осадження [теоретично] неможливо!
Будь-який процес, в якому утворюється нова фаза в межах існуючої однорідної фази, стикається з проблемою зародкування: найменші з цих нових фаз - краплі дощу, що утворюються в повітрі, крихітні бульбашки, що утворюються в рідині при температурі кипіння - за своєю суттю менш стабільні, ніж більші, і тому схильні зникати. Те ж саме стосується утворення осаду: якщо більш дрібні кристали більш розчинні, то як взагалі може утворитися найдрібніший, перший кристал?
У будь-якому іонному розчині звичайні колізійні процеси постійно утворюються невеликі скупчення протилежно заряджених іонів. Найменші з цих агрегатів володіють більш високою вільною енергією, ніж ізольовані сольватние іони, і вони швидко дисоціюють. Іноді, однак, один з цих протокристалітів досягає критичного розміру, стабільність якого дозволяє йому залишатися цілим досить довго, щоб служити поверхнею («ядром»), на яку осадження додаткових іонів може призвести до ще більшої стабільності. У цей момент процес переходить від зародження до стадії росту.
Теоретичні розрахунки прогнозують, що зародження з ідеально однорідного розчину є досить малоймовірним процесом; десятикратне перенасичення повинно виробляти тільки одне ядро на см 3 в рік. Тому вважається, що більшість зародження відбувається неоднорідно на поверхні якоїсь іншої частинки, можливо, частинки пилу. Ефективність цього процесу критично залежить від характеру і стану поверхні, що дає початок ядру.