9.4: Окислювально-відновне титрування
- Page ID
- 24856

Аналітичні титрування з використанням окислювально-відновних реакцій були введені незабаром після розробки кислотно-лужної титріметрії. Найбільш раннє окислювально-відновне титрування скористалося окислювальною силою хлору. У 1787 році Клод Бертоле ввів метод кількісного аналізу хлорної води (суміші Cl 2, HCl і HoCl), заснований на її здатності окислювати індиго - барвник, безбарвний в окисленому стані. У 1814 році Джозеф Гей-Люссак розробив подібний метод визначення хлору в відбілюючому порошку. В обох методах кінцевою точкою є зміна кольору. До точки еквівалентності розчин безбарвний через окислення індиго. Після точки еквівалентності, однак, не відреагував індиго надає постійний колір розчину.
Кількість окислювально-титриметричних методів збільшилася в середині 1800-х років з введенням\(\text{MnO}_4^-\)\(\text{Cr}_2\text{O}_7^{2-}\), і I 2 як окислювальних титрантів, і Fe 2 + і\(\text{S}_2\text{O}_3^{2-}\) як відновлювальних титрантів. Навіть при наявності цих нових титрантів окислювально-відновлювально-відновлювальна титриметрія розвивалася повільно через відсутність відповідних показників. Титрант може служити власним показником, якщо його окислені і відновлені форми значно відрізняються за кольором. Наприклад, інтенсивно фіолетовий\(\text{MnO}_4^-\) іон служить власним індикатором, оскільки його зменшена форма, Mn 2 +, майже безбарвна. Інші титранти вимагають окремого показника. Перший такий показник, дифеніламін, був введений в 1920-х роках. Незабаром послідували інші окислювально-відновні показники, збільшуючи застосовність окислювально-відновної титриметрії.
Криві окислювально-відновного титрування
Щоб оцінити окислювально-відновне титрування, нам потрібно знати форму кривої титрування. При кислотно-луговому титруванні або титруванні комплексоутворення крива титрування показує, як змінюється концентрація H 3 O + (як рН) або M n + (як pM), коли ми додаємо титрант. Для окислювально-відновного титрування зручно контролювати потенціал реакції титрування замість концентрації одного виду.
Ви можете згадати з глави 6, що рівняння Нернста пов'язує потенціал розчину з концентраціями реагентів і продуктів, які беруть участь у окислювально-відновній реакції. Розглянемо, наприклад, титранд, при якому титранд в відновленому стані, А червоний, вступає в реакцію з титрантом в окисленому стані, В бокс.
\[A_{red} + B_{ox} \rightleftharpoons B_{red} + A_{ox} \nonumber\]
де А вол - окислена форма титранду, B червоний - відновлена форма титранту, а стехіометрія між ними - 1:1. Потенціал реакції, E rxn, - це різниця між потенціалами відновлення для кожної половини реакції.
\[E_{rxn} = E_{B_{ox}/B_{red}} - E_{A_{ox}/A_{red}} \nonumber\]
Після кожного додавання титранту реакція між титрандом і титрантом досягає стану рівноваги. Оскільки потенціал при рівновазі дорівнює нулю, потенціали відновлення титранду та титранта ідентичні.
\[E_{B_{ox}/B_{red}} = E_{A_{ox}/A_{red}} \nonumber\]
Це важливе спостереження, оскільки дозволяє нам використовувати або половину реакції для моніторингу прогресу титрування.
Перед точкою еквівалентності титрувальна суміш складається з помітних кількостей окисленої та відновленої форм титранду. Концентрація не прореагував титранту, однак, дуже мала. Отже, потенціал легше обчислити, якщо використовувати рівняння Нернста для напівреакції титранда
\[E_{rxn} = E_{A_{ox}/A_{red}}^{\circ} - \frac{RT}{nF}\ln{\frac{[A_{red}]}{[A_{ox}]}} \nonumber\]
Після точки еквівалентності легше обчислити потенціал, використовуючи рівняння Нернста для напівреакції титранта.
\[E_{rxn} = E_{B_{ox}/B_{red}}^{\circ} - \frac{RT}{nF}\ln{\frac{[B_{red}]}{[B_{ox}]}} \nonumber\]
Хоча рівняння Нернста записано через стандартний потенціал стану напівреакції, замість нього часто використовується формальний потенціал, залежний від матриці. Див. Додаток 13 для стандартних потенціалів стану та формальних потенціалів для окремих напівреакцій.
Розрахунок кривої титрування
Розрахуємо криву титрування для титрування 50,0 мл 0,100 M Fe 2 + з 0.100 M Ce 4 + в матриці 1 M HClO 4. Реакція в даному випадку така:
Оскільки константа рівноваги для реакції\ ref {9.1} дуже велика - це приблизно\(6 \times 10^{15}\) —можна припустити, що аналіт і титрант реагують повністю.
У 1 М HClO 4 формальний потенціал зниження Fe 3 + до Fe 2 + становить +0,767 В, а формальний потенціал зниження Ce 4 + до Ce 3 + становить +1,70 В.
Перше завдання полягає в тому, щоб обчислити обсяг Ce, необхідний для досягнення точки еквівалентності титрування. З стехіометрії реакції ми знаємо, що
\[\text{mol Fe}^{2+} = M_\text{Fe}V_\text{Fe} = M_\text{Ce}V_\text{Ce} = \text{mol Ce}^{4+} \nonumber\]
Розв'язування для об'єму Ce 4 + дає точковий об'єм еквівалентності як
\[V_{eq} = V_\text{Ce} = \frac{M_\text{Fe}V_\text{Fe}}{M_\text{Ce}} = \frac{(0.100 \text{ M})(50.0 \text{ mL})}{(0.100 \text{ M})} = 50.0 \text{ mL} \nonumber\]
До точки еквівалентності концентрацію не прореагував Fe 2 + і концентрацію Fe 3 + розрахувати нескладно. З цієї причини ми знаходимо потенціал за допомогою рівняння Нернста для напівреакції Fe 3 +/Fe 2 +.
\[E = +0.767 \text{ V} - 0.05916 \log{\frac{[\text{Fe}^{2+}]}{[\text{Fe}^{3+}]}} \label{9.2}\]
Наприклад, концентрації Fe 2 + і Fe 3 + після додавання 10,0 мл титранту становлять
\[[\text{Fe}^{2+}] = \frac{(\text{mol Fe}^{2+})_\text{initial} - (\text{mol Ce}^{4+})_\text{added}}{\text{total volume}} = \frac{M_\text{Fe}V_\text{Fe} - M_\text{Ce}V_\text{Ce}}{V_\text{Fe} + V_\text{Ce}} \nonumber\]
\[[\text{Fe}^{2+}] = \frac{(0.100 \text{ M})(50.0 \text{ mL}) - (0.100 \text{ M})(10.0 \text{ mL})}{50.0 \text{ mL} + 10.0 \text{ mL}} = 6.67 \times 10^{-2} \text{ M} \nonumber\]
\[[\text{Fe}^{3+}] = \frac{(\text{mol Ce}^{4+})_\text{added}}{\text{total volume}} = \frac{M_\text{Ce}V_\text{Ce}}{V_\text{Fe} + V_\text{Ce}} \nonumber\]
\[[\text{Fe}^{3+}] = \frac{(0.100 \text{ M})(10.0 \text{ mL})}{50.0 \text{ mL} + 10.0 \text{ mL}} = 1.67 \times 10^{-2} \text{ M} \nonumber\]
Підстановка цих концентрацій на Equation\ ref {9.2} дає потенціал як
\[E = +0.767 \text{ V} - 0.05916 \log{\frac{6.67 \times 10^{-2}}{1.67 \times 10^{-2}}} = +0.731 \text{ V} \nonumber\]
Після точки еквівалентності концентрацію Се 3 + і концентрацію надлишку Се 4 + розрахувати нескладно. З цієї причини ми знаходимо потенціал, використовуючи рівняння Нернста для напівреакції Ce 4 + +/Ce 3+ подібним до того, який використовувався вище для обчислення потенціалів перед точкою еквівалентності.
\[E = +1.70 \text{ V} - 0.05916 \log{\frac{[\text{Ce}^{3+}]}{[\text{Ce}^{4+}]}} \label{9.3}\]
Наприклад, після додавання 60,0 мл титранту концентрації Ce 3 + і Ce 4 + складають
\[[\text{Ce}^{3+}] = \frac{(\text{mol Fe}^{2+})_\text{initial}}{\text{total volume}} = \frac{M_\text{Fe}V_\text{Fe}}{V_\text{Fe}+V_\text{Ce}} \nonumber\]
\[[\text{Ce}^{3+}] = \frac{(0.100 \text{ M})(50.0 \text{ mL})}{50.0 \text{ mL} + 60.0 \text{ mL}} = 4.55 \times 10^{-2} \text{ M} \nonumber\]
\[[\text{Ce}^{4+}] = \frac{(\text{mol Ce}^{4+})_\text{added}-(\text{mol Fe}^{2+})_\text{initial}}{\text{total volume}} = \frac{M_\text{Ce}V_\text{Ce}-M_\text{Fe}V_\text{Fe}}{V_\text{Fe}+V_\text{Ce}} \nonumber\]
\[[\text{Ce}^{4+}] = \frac{(0.100 \text{ M})(60.0 \text{ mL})-(0.100 \text{ M})(50.0 \text{ mL})}{50.0 \text{ mL} + 60.0 \text{ mL}} = 9.09 \times 10^{-3} \text{ M} \nonumber\]
Підстановка цих концентрацій у рівняння\ ref {9.3} дає потенціал
\[E = +1.70 \text{ V} - 0.05916 \log{\frac{4.55 \times 10^{-2} \text{ M}}{9.09 \times 10^{-3} \text{ M}}} = +1.66 \text{ V} \nonumber\]
У точці еквівалентності титрування потенціал, E eq, у Equation\ ref {9.2} та Equation\ ref {9.3} ідентичні. Додавання рівнянь разом до дає
\[2E_{eq} = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} - 0.05916 \log{\frac{[\text{Fe}^{2+}][\text{Ce}^{3+}]}{[\text{Fe}^{3+}][\text{Ce}^{4+}]}} \nonumber\]
Оскільки [Fe 2 +] = [Ce 4 +] і [Ce 3 +] = [Fe 3+] в точці еквівалентності, термін журналу має значення нуль, а потенціал точки еквівалентності дорівнює
\[E_{eq} = \frac{E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ}}{2} = \frac{0.767 \text{ V} + 1.70 \text{ V}}{2} = +1.23 \text{ V} \nonumber\]
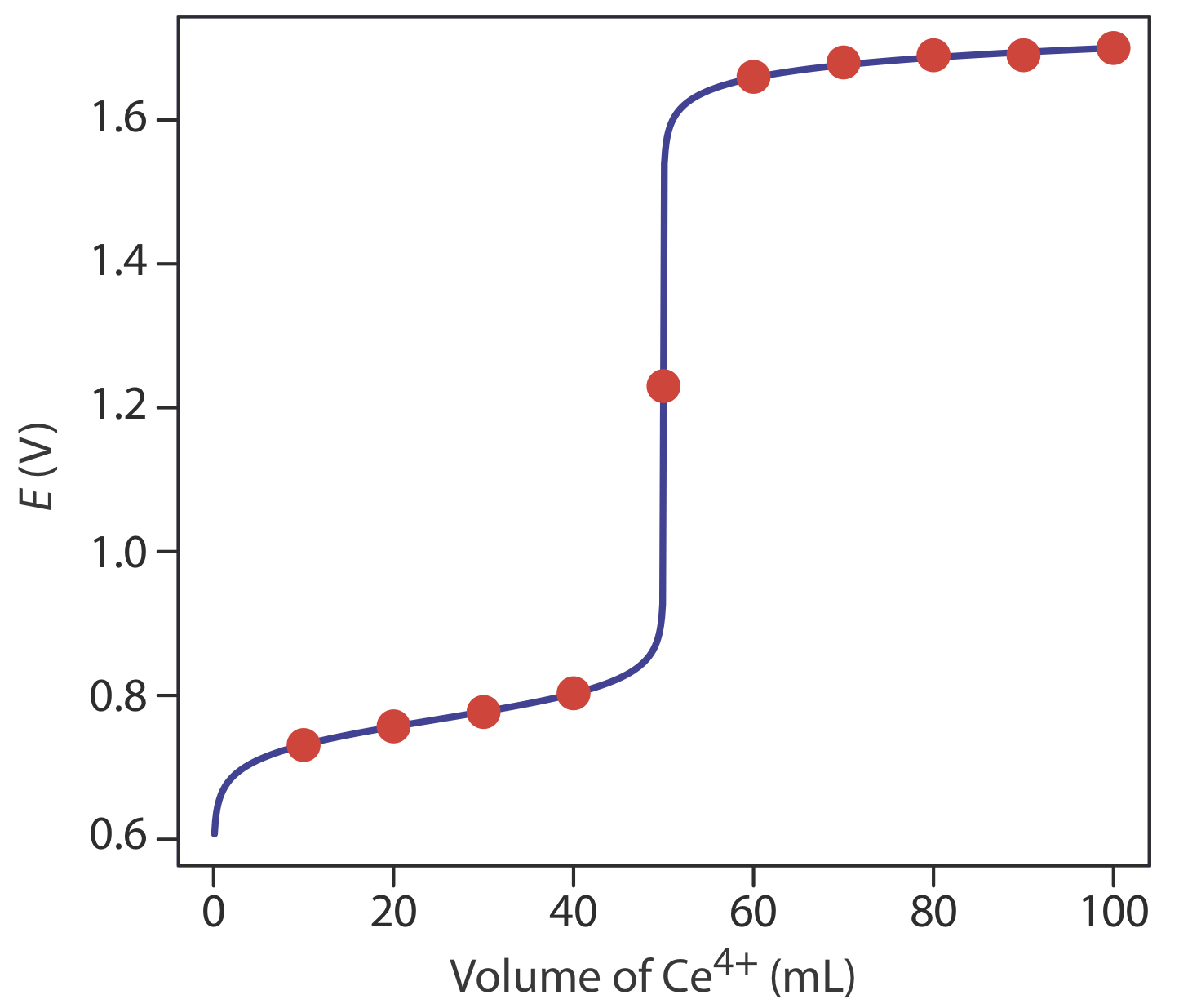
Додаткові результати для цієї кривої титрування наведено у таблиці Template:index та рисунку Template:index.
| Обсяг Се 4 + (мл) | Е (V) | Обсяг Се 4 + (мл) | Е (V) |
|---|---|---|---|
| 10.0 | 0,731 | 60.0 | 1.66 |
| 20.0 | 0,757 | 70.0 | 1.68 |
| 30.0 | 0.777 | 80.0 | 1.69 |
| 40.0 | 0.803 | 90.0 | 1.69 |
| 50.0 | 1.23 | 100.0 | 1,70 |
Обчисліть криву титрування для титрування 50,0 мл 0,0500 M Sn 2 + з 0.100 M Tl 3 +. І титранд, і титрант становлять 1,0 М в HCl. Реакція титрування є
\[\text{Sn}^{2+}(aq) + \text{Tl}^{3+} \rightleftharpoons \text{Tl}^+(aq) + \text{Sn}^{4+}(aq) \nonumber\]
- Відповідь
-
Обсяг Tl 3 +, необхідний для досягнення точки еквівалентності
\[V_{eq} = V_\text{Tl} = \frac{M_\text{Sn}V_\text{Sn}}{M_\text{Tl}} = \frac{(0.050 \text{ M})(50.0 \text{ mL})}{(0.100 \text{ M})} = 25.0 \text{ mL} \nonumber\]
До точки еквівалентності концентрацію не прореагував Sn 2 + і концентрацію Sn 4 + розрахувати нескладно. З цієї причини ми знаходимо потенціал, використовуючи рівняння Нернста для напівреакції Sn 4 + + /Sn 2+. Наприклад, концентрації Sn 2 + і Sn 4 + після додавання 10,0 мл титранту становлять
\[[\text{Sn}^{2+}] = \frac{(0.050 \text{ M})(50.0 \text{ mL}) - (0.100 \text{ M})(10.0 \text{ mL})}{50.0 \text{ mL} + 10.0 \text{ mL}} = 0.0250 \text{ M} \nonumber\]
\[[\text{Sn}^{4+}] = \frac{(0.100 \text{ M})(10.0 \text{ mL})}{50.0 \text{ mL} + 10.0 \text{ mL}} = 0.0167 \text{ M} \nonumber\]
і потенціал є
\[E = +0.139 \text{ V} - \frac{0.05916}{2} \log{\frac{0.0250 \text{ M}}{0.0167 \text{ M}}} = +0.134 \text{ V} \nonumber\]
Після точки еквівалентності концентрацію Tl + і концентрацію надлишку Tl 3 + обчислити нескладно. З цієї причини ми знаходимо потенціал за допомогою рівняння Нернста для напівреакції Tl 3 + /Tl +. Наприклад, після додавання 40,0 мл титранту концентрації Tl + і Tl 3 + складають
\[[\text{Tl}^{+}] = \frac{(0.050 \text{ M})(50.0 \text{ mL})}{50.0 \text{ mL} + 40.0 \text{ mL}} = 0.0278 \text{ M} \nonumber\]
\[[\text{Tl}^{3+}] = \frac{(0.100 \text{ M})(40.0 \text{ mL}) - (0.050 \text{ M})(50.0 \text{ mL})}{50.0 \text{ mL} + 40.0 \text{ mL}} = 0.0167 \text{ M} \nonumber\]
і потенціал є
\[E = +0.77 \text{ V} - \frac{0.05916}{2} \log{\frac{0.0278 \text{ M}}{0.0167 \text{ M}}} = +0.76 \text{ V} \nonumber\]
У точці еквівалентності титрування потенціал, E екв, потенціал дорівнює
\[E_{eq} = \frac{0.139 \text{ V} + 0.77 \text{ V}}{2} = +0.45 \text{ V} \nonumber\]
Деякі додаткові результати наведені тут.
Обсяг Tl 3 + (мл) Е (V) Обсяг Tl 3 + (мл) Е (V) 5.00 0,121 30.0 0,75 10.0 0.134 35.0 0,75 15,0 0.144 40.0 0,76 20.0 0.157 45.0 0,76 25.0 0,45
Ескіз кривої окислювально-відновного титрування
Для оцінки співвідношення між точкою еквівалентності титрування та її кінцевою точкою потрібно побудувати лише розумне наближення точної кривої титрування. У цьому розділі ми продемонструємо простий метод ескізу кривої окислювально-відновного титрування. Наша мета - швидко намалювати криву титрування, використовуючи якомога менше розрахунків. Скористаємося титруванням 50,0 мл 0.100 M Fe 2 + з 0.100 M Ce 4 + в матриці 1 M HClO 4.
Це той самий приклад, який ми використовували при розробці розрахунків для кривої окислювально-відновного титрування. Ви можете переглянути результати цього розрахунку у таблиці Template:index та на малюнку Template:index.
Почнемо з розрахунку об'єму точки еквівалентності титрування, який, як ми визначили раніше, становить 50,0 мл. Далі ми малюємо наші осі, розмістивши потенціал, E, на осі y і об'єм титранта на осі x. Щоб вказати об'єм точки еквівалентності, проведемо вертикальну лінію, яка перетинає вісь x на 50,0 мл Ce 4 +. Рисунок Template:index a показує результат першого кроку нашого ескізу.
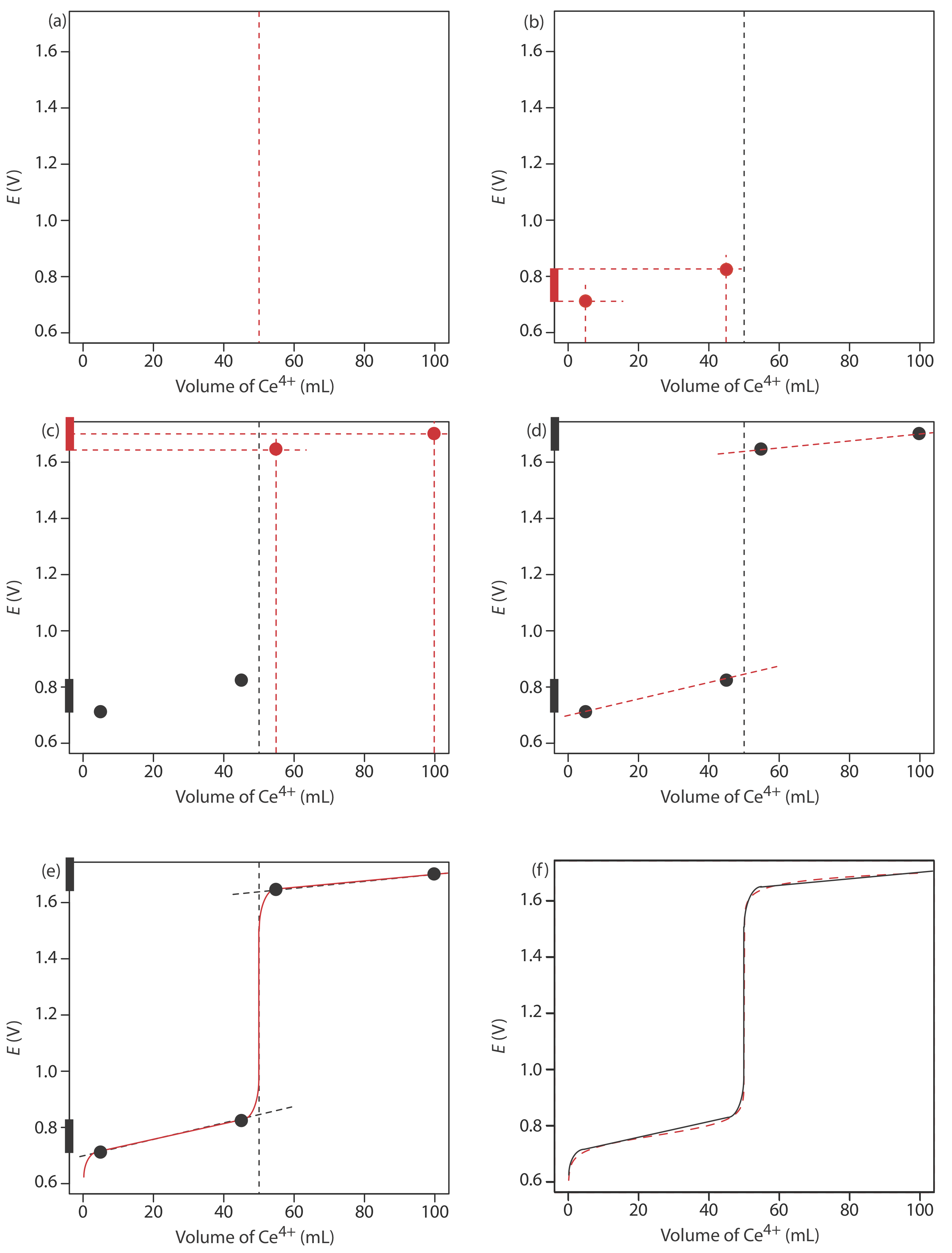
Перед точкою еквівалентності потенціал визначається окисно-відновним буфером Fe 2 + і Fe 3 +. Хоча ми можемо обчислити потенціал за допомогою рівняння Нернста, ми можемо уникнути цього розрахунку, якщо зробити просте припущення. Ви можете згадати з глави 6, що окислювально-відновний буфер працює над діапазоном потенціалів, який поширюється приблизно ± (0,05916/n) одиниця з обох боків\(E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ}\). Потенціал на нижній межі буфера дорівнює
\[E = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} - 0.05916 \nonumber\]
коли концентрація Fe 2 +\(10 \times\) більше, ніж у Fe 3 +. Буфер досягає свого верхнього потенціалу
\[E = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 0.05916 \nonumber\]
коли концентрація Fe 2 +\(10 \times\) менше, ніж у Fe 3 +. Окислювально-відновний буфер охоплює діапазон обсягів від приблизно 10% об'єму точки еквівалентності до приблизно 90% об'єму точки еквівалентності. На малюнку Template:index b показано другий крок нашого ескізу. Спочатку ми накладаємо сходову діаграму для Fe на вісь y, використовуючи її\(E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ}\) значення 0.767 V і включаючи діапазон потенціалів буфера. Далі додаємо бали для потенціалу при 10% V екв (потенціал 0,708 В при 5,0 мл) і для потенціалу при 90% V екв (потенціал 0,826 В при 45,0 мл).
Аналогічний підхід ми використовували при накресленні кривої титрування кислота-основа для титрування оцтової кислоти з NaOH; докладніше див. Розділ 9.2.
Третім кроком ескізу нашої кривої титрування є додавання двох точок після точки еквівалентності. Тут потенціал контролюється окисно-відновним буфером Ce 3 + і Ce 4 +. Окислювально-відновний буфер знаходиться на нижній межі
\[E = E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} - 0.05916 \nonumber\]
коли титрант досягає 110% об'єму точки еквівалентності, а потенціал -\(E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ}\) коли об'єм Ce дорівнює\(2 \times V_{eq}\).
Аналогічний підхід ми використовували при накресленні кривої титрування комплексоутворення для титрування Mg 2 + за допомогою ЕДТА; докладніше див. Розділ 9.3.
Рисунок Template:index c показує третій крок нашого ескізу. Спочатку ми накладаємо діаграму сходів для Ce на вісь y, використовуючи її\(E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ}\) значення 1.70 V і включаючи діапазон буфера. Далі додаємо точки, що представляють потенціал при 110% V екв (значення 1,66 В при 55,0 мл) і при 200% V екв (значення 1,70 В при 100,0 мл).
Далі проводимо пряму лінію через кожну пару точок, продовжуючи лінію через вертикальну лінію, яка вказує на об'єм точки еквівалентності (Рисунок Template:index d). Нарешті, ми завершуємо наш ескіз, намалювавши плавну криву, яка з'єднує три прямолінійні відрізки (Рисунок Template:index e). Порівняння нашого ескізу з точною кривою титрування (Рисунок Template:index f) показує, що вони знаходяться в тісній згоді.
Намалюйте криву титрування для титрування 50,0 мл 0,0500 M Sn 4 + з 0.100 M Tl +. І титранд, і титрант становлять 1,0 М в HCl. Реакція титрування є
\[\text{Sn}^{2+}(aq) + \text{Tl}^{3+}(aq) \rightleftharpoons \text{Tl}^{+}(aq) + \text{Sn}^{4+}(aq) \nonumber\]
Порівняйте свій ескіз з обчисленою кривою титрування з вправи Template:index.
- Відповідь
-
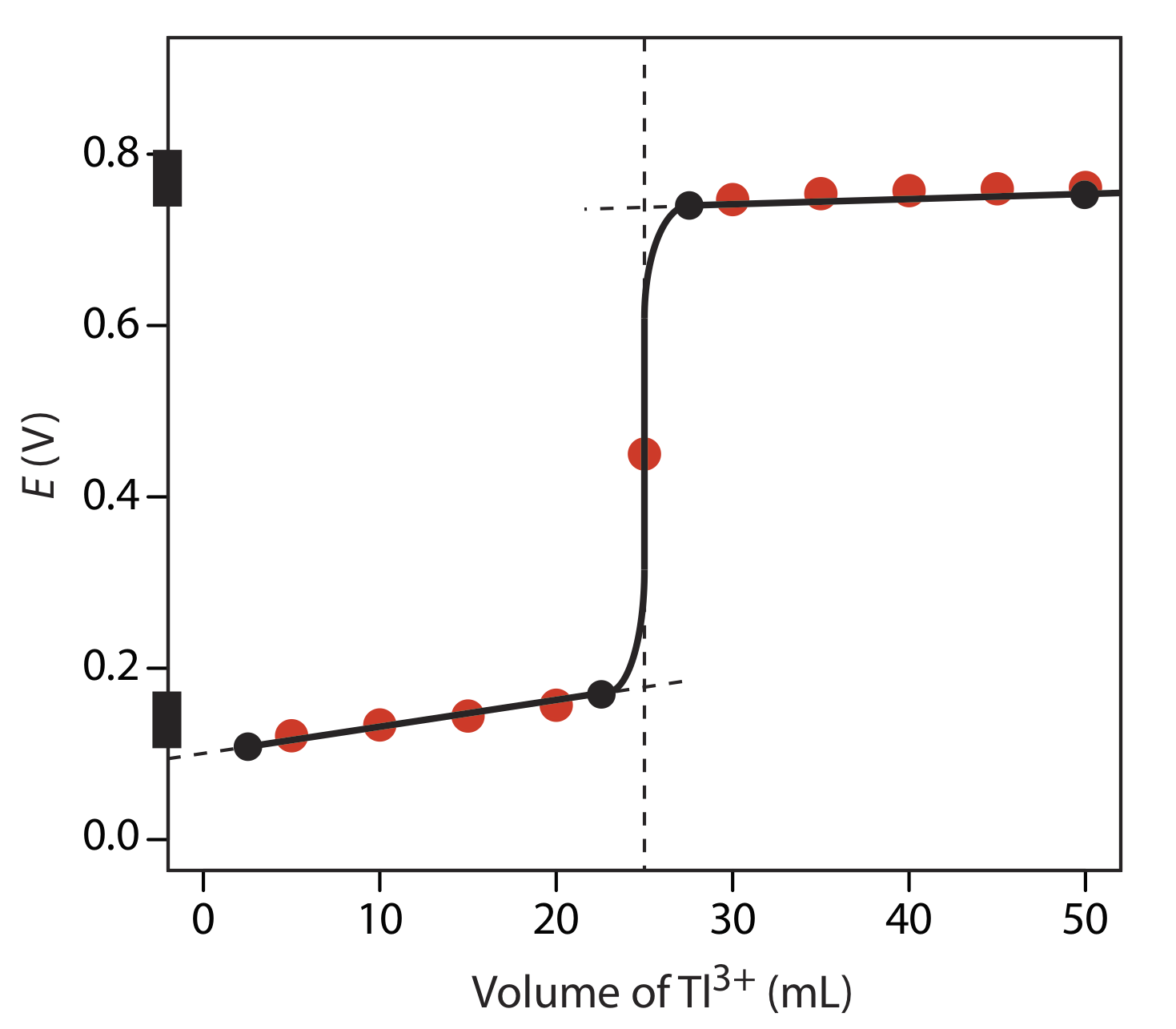
На малюнку нижче показаний ескіз кривої титрування. Дві точки перед точкою еквівалентності
V Тл = 2,5 мл, Е = +0,109 В і V Тл = 22,5 мл, Е = +0,169 В
побудовані за допомогою окислювально-відновного буфера для Sn 4 +/Sn 2+, який охоплює потенційний діапазон +0,139 ± 0,5916/2. Дві точки після точки еквівалентності
V Тл = 27,5 мл, Е = +0,74 В і V Тл = 50 мл, Е = +0,77 В
побудовані за допомогою окислювально-відновного буфера для Tl 3 + /Tl +, який охоплює діапазон потенціалів +0,139 ± 0,5916/2. Чорні точки і крива є приблизним ескізом кривої титрування. Точки червоного кольору - це обчислення з вправи Template:index.
Вибір та оцінка кінцевої точки
Точка еквівалентності окислювально-відновного титрування виникає, коли ми реагуємо стехіометрично еквівалентні кількості титранду та титранту. Як і у випадку кислотно-лужних титрувань та титрування комплексоутворення, оцінено точку еквівалентності окислювально-відновного титрування з використанням експериментальної кінцевої точки. Для визначення кінцевої точки окислювально-відновного титрування доступні різні методи, включаючи індикатори та датчики, які реагують на зміну умов розчину.
Де знаходиться точка еквівалентності
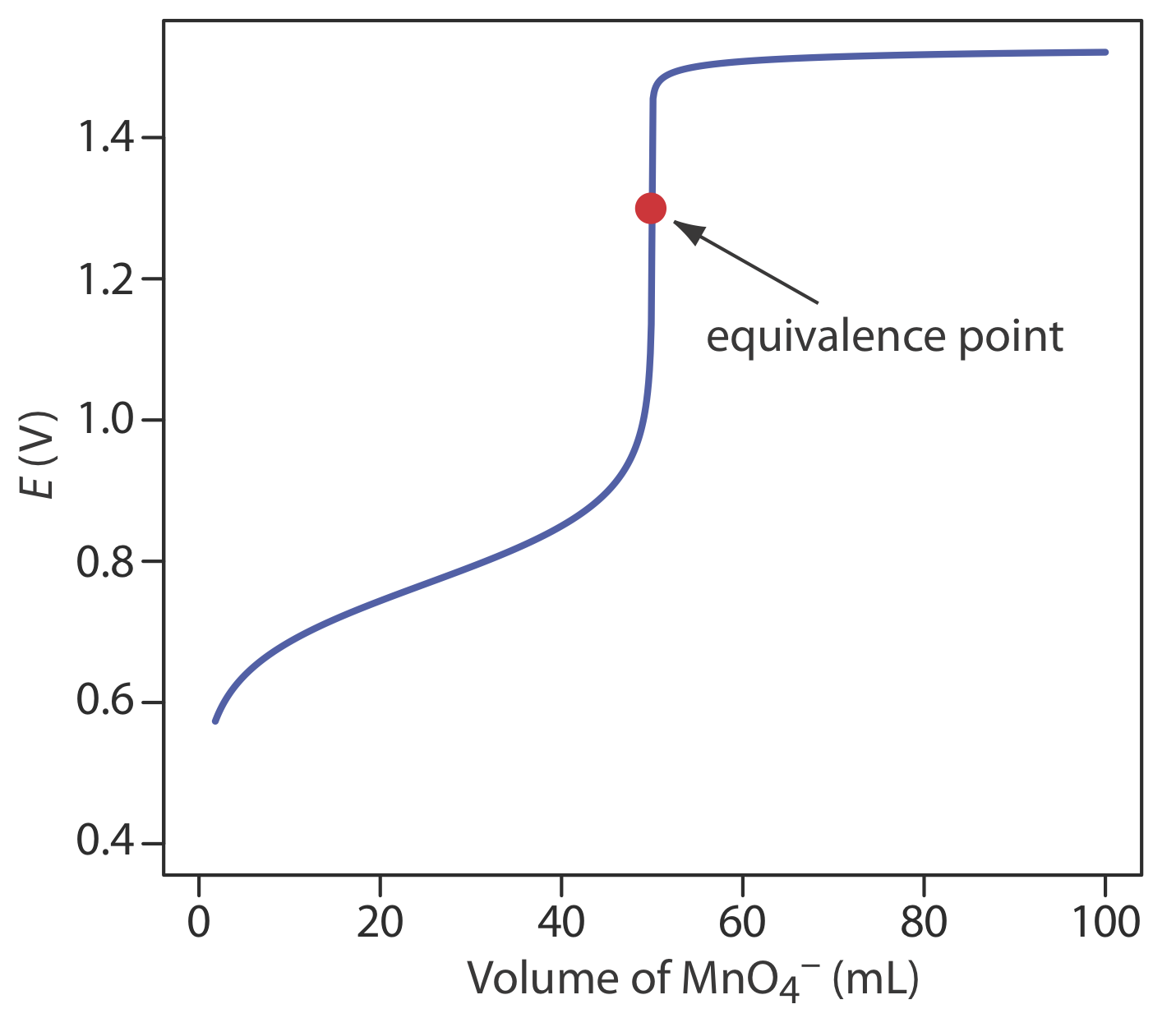
Для кислотно-основного титрування або комплексометричного титрування точка еквівалентності майже ідентична точці перегину на круто зростаючій частині кривої титрування. Якщо ви озирнетеся назад на рисунок 9.2.2 та рисунок 9.3.3, ви побачите, що точка перегину знаходиться посередині цього крутого підйому кривої титрування, що дозволяє відносно легко знайти точку еквівалентності під час ескізу цих кривих титрування. Ми називаємо це симетричною точкою еквівалентності. Якщо стехіометрія окислювально-відновного титрування становить 1:1 - тобто один моль титранту реагує з кожним молем титранду - тоді точка еквівалентності симетрична. Якщо стехіометрія реакції титрування не дорівнює 1:1, то точка еквівалентності ближче до верхньої або нижньої частини різкого підйому кривої титрування. У цьому випадку ми маємо асиметричну точку еквівалентності.
Вивести загальне рівняння потенціалу точки еквівалентності при титруванні Fe 2 + с\(\text{MnO}_4^-\).
\[5\text{Fe}^{2+}(aq) + \text{MnO}_4^-(aq) + 8\text{H}^+(aq) \rightarrow 5\text{Fe}^{3+}(aq) + \text{Mn}^{2+}(aq) + 4\text{H}_2\text{O}(l) \nonumber\]
Рішення
Напівреакції на окислення Fe 2 + і відновлення\(\text{MnO}_4^-\) є
\[\text{Fe}^{2+}(aq) \rightarrow \text{Fe}^{3+}(aq) + e^- \nonumber\]
\[\text{MnO}_4^-(aq) + 8\text{H}^+(aq) + 5 e^- \rightarrow \text{Mn}^{2+}(aq) + 4\text{H}_2\text{O}(l) \nonumber\]
для яких рівняння Нернста є
\[E = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} - 0.5916 \log{\frac{[\text{Fe}^{2+}]}{[\text{Fe}^{3+}]}} \nonumber\]
\[E = E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ} - \frac{0.5916}{5} \log{\frac{[\text{Mn}^{2+}]}{[\text{MnO}_4^{-}][\text{H}^+]^8}} \nonumber\]
Перш ніж скласти ці два рівняння, ми повинні помножити друге рівняння на 5, щоб ми могли об'єднати терміни журналу; таким чином
\[6E_{eq} = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 5E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ} - 0.05916 \log{\frac{[\text{Fe}^{2+}][\text{Mn}^{2+}]}{[\text{Fe}^{3+}][\text{MnO}_4^{-}][\text{H}^+]^8}} \nonumber\]
У точці еквівалентності ми знаємо, що
\[[\text{Fe}^{2+}] = 5 \times [\text{MnO}_4^-] \text{ and } [\text{Fe}^{3+}] = 5 \times [\text{Mn}^{2+}] \nonumber\]
Підстановка цих рівнянь у попереднє рівняння та перестановка дає нам загальне рівняння потенціалу в точці еквівалентності.
\[6E_{eq} = E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 5E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ} - 0.05916 \log{\frac{5[\text{MnO}_4^{-}][\text{Mn}^{2+}]}{5[\text{Mn}^{2+}][\text{MnO}_4^{-}][\text{H}^+]^8}} \nonumber\]
\[E_{eq} = \frac{E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 5E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ}}{6} - \frac{0.05916}{6} \log{\frac{1}{[\text{H}^+]^8}} \nonumber\]
\[E_{eq} = \frac{E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 5E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ}}{6} + \frac{0.05916 \times 8}{6} \log{[\text{H}^+]} \nonumber\]
\[E_{eq} = \frac{E_{\text{Fe}^{3+}/\text{Fe}^{2+}}^{\circ} + 5E_{\text{MnO}_4^{-}/\text{Mn}^{2+}}^{\circ}}{6} - 0.07888 \text{pH} \nonumber\]
Наше рівняння для точки еквівалентності має два домені. Перший член - це середньозважене значення потенціалів стандартного стану титрану та титранту, в якому ваговими факторами є кількість електронів у відповідних напівреакціях. Другий термін показує, що E eq для цього титрування залежить від рН. При рН 1 (в H 2 SO 4), наприклад, точка еквівалентності має потенціал
\[E_{eq} = \frac{0.768 + 5 \times 1.51}{6} - 0.07888 \times 1 = 1.31 \text{ V} \nonumber\]
На малюнку Template:index показана типова крива титрування для титрування Fe 2 + с\(\text{MnO}_4^-\). Зверніть увагу, що точка еквівалентності титрування асиметрична.
Вивести загальне рівняння потенціалу точки еквівалентності для титрування U 4+ з Ce 4 +. Незбалансована реакція є
\[\text{Ce}^{4+}(aq) + \text{U}^{4+}(aq) \rightarrow \text{UO}_2^{2+}(aq) + \text{Ce}^{3+}(aq) \nonumber\]
Який потенціал точки еквівалентності, якщо рН дорівнює 1?
- Відповідь
-
Дві половинні реакції
\[\text{Ce}^{4+}(aq) + e^- \rightarrow \text{Ce}^{3+}(aq) \nonumber\]
\[\text{U}^{4+}(aq) +2\text{H}_2\text{O}(l) \rightarrow \text{UO}_2^{2+}(aq)) + 4\text{H}^+(aq) +2e^- \nonumber\]
для яких рівняння Нернста є
\[E = E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} - 0.05916 \log{\frac{[\text{Ce}^{3+}]}{[\text{Ce}^{4+}]}} \nonumber\]
\[E = E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ} - \frac{0.05916}{2} \log{\frac{[\text{U}^{4+}]}{[\text{UO}_2^{2+}][\text{H}^+]^4}} \nonumber\]
Перш ніж скласти ці два рівняння разом, ми повинні помножити друге рівняння на 2, щоб ми могли об'єднати терміни журналу; таким чином
\[3E = E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} + 2E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ} - 0.05916 \log{\frac{[\text{Ce}^{3+}][\text{U}^{4+}]}{[\text{Ce}^{4+}][\text{UO}_2^{2+}][\text{H}^+]^4}} \nonumber\]
У точці еквівалентності ми знаємо, що
\[[\text{Ce}^{3+}] = 2 \times [\text{UO}_2^{2+}] \text{ and } [\text{Ce}^{4+}] = 2 \times [\text{U}^{4+}] \nonumber\]
Підстановка цих рівнянь у попереднє рівняння та перестановка дає нам загальне рівняння потенціалу в точці еквівалентності.
\[3E = E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} + 2E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ} - 0.05916 \log{\frac{2[\text{UO}_2^{2+}][\text{U}^{4+}]}{2[\text{U}^{4+}][\text{UO}_2^{2+}][\text{H}^+]^4}} \nonumber\]
\[E = \frac{E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} + 2E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ}}{3} - \frac{0.05916}{3} \log{\frac{1}{[\text{H}^+]^4}} \nonumber\]
\[E = \frac{E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} + 2E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ}}{3} + \frac{0.05916 \times 4}{3} \log{[\text{H}^+]^4} \nonumber\]
\[E = \frac{E_{\text{Ce}^{4+}/\text{Ce}^{3+}}^{\circ} + 2E_{\text{UO}_2^{2+}/\text{U}^{4+}}^{\circ}}{3} - 0.07888\text{pH} \nonumber\]
При рН 1 точка еквівалентності має потенціал
\[E = \frac{1.72 + 2 \times 0.327}{3} - 0.07888 \times 1 = +0.712 \text{ V} \nonumber\]
Пошук кінцевої точки за допомогою індикатора
Три типи індикаторів використовуються для сигналізації кінцевої точки окислювально-відновного титрування. Окислені і відновлені форми деяких титрантів, наприклад\(\text{MnO}_4^-\), мають різні кольори. Розчин\(\text{MnO}_4^-\) інтенсивно фіолетового кольору. Однак у кислому розчині відновлена форма перманганату, Mn 2 +, майже безбарвна. При використанні в\(\text{MnO}_4^-\) якості титранта розчин титранда залишається безбарвним до точки еквівалентності. Перша крапля надлишку\(\text{MnO}_4^-\) виробляє постійний відтінок фіолетового кольору, сигналізуючи про кінцеву точку.
Деякі показники утворюють кольорове з'єднання зі специфічною окисленою або відновленою формою титранта або титранда. Крохмаль, наприклад, утворює темно-фіолетовий комплекс с\(\text{I}_3^-\). Ми можемо використовувати цей чіткий колір, щоб сигналізувати про наявність надлишку\(\text{I}_3^-\) як титранта - зміна кольору від безбарвного до фіолетового - або завершення реакції, яка споживає\(\text{I}_3^-\) як титран - зміна кольору від фіолетового до безбарвного. Іншим прикладом специфічного показника є тіоцианат SCN —, який утворює розчинний комплекс червоного кольору Fe (SCN) 2+ в присутності Fe 3 +.
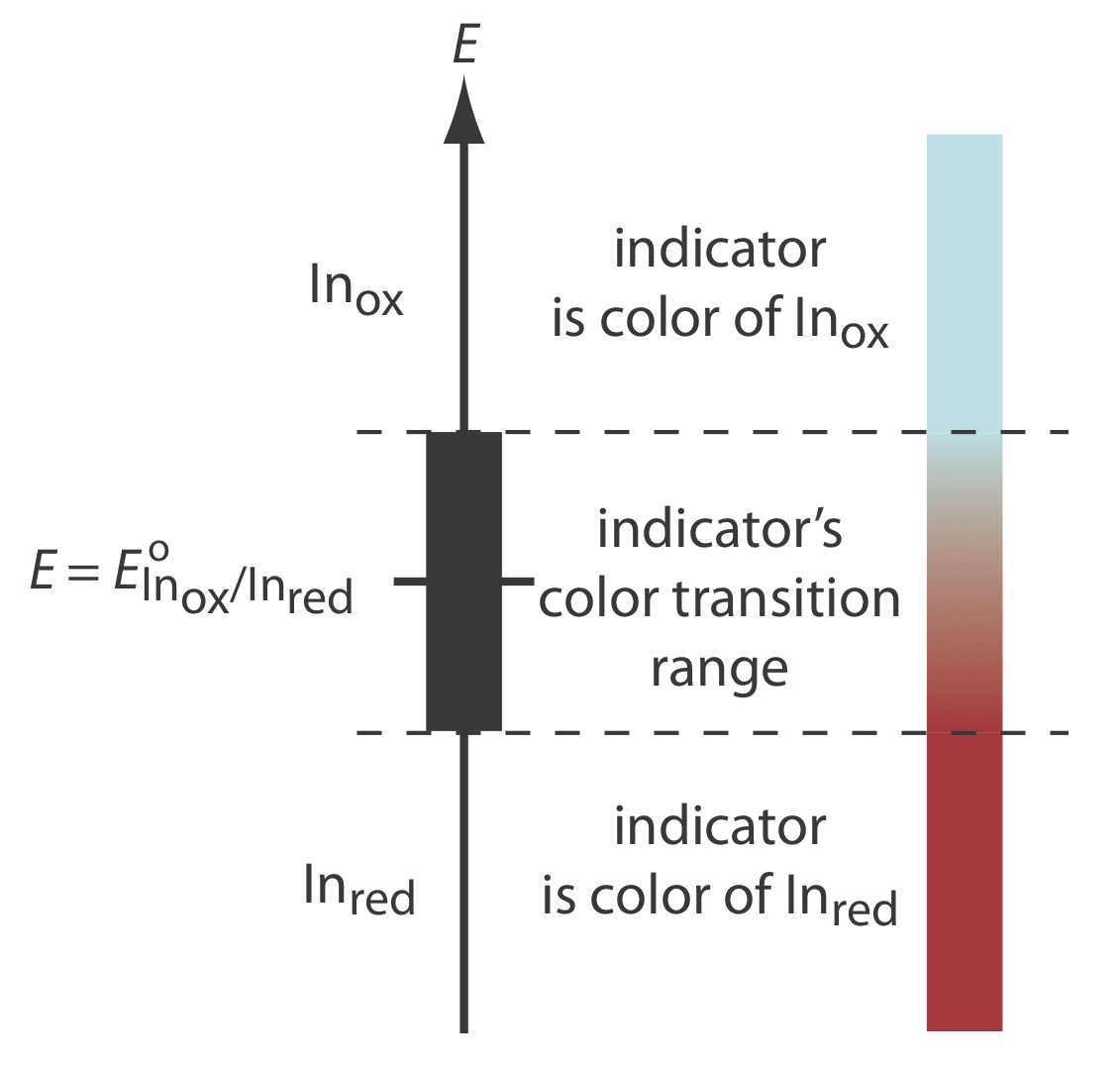
Найважливішим класом показників є речовини, які не беруть участі в окислювально-відновному титруванні, але окислені і відновлені форми яких відрізняються за кольором. Коли ми додаємо окислювально-відновний індикатор до титранду, індикатор надає колір, який залежить від потенціалу рішення. У міру зміни потенціалу розчину з додаванням титранту індикатор з часом змінює ступінь окислення і змінює колір, сигналізуючи про кінцеву точку.
Щоб зрозуміти взаємозв'язок між потенціалом і кольором індикатора, розглянемо його редукційну напівреакцію
\[\text{In}_\text{ox} + ne^- \rightleftharpoons \text{In}_\text{red} \nonumber\]
де In box і In red - це, відповідно, окислена і відновлена форми індикатора.
Для простоти, У вола і В червоному показані без конкретних зарядів. Оскільки відбувається зміна ступеня окислення, In box і In red не можуть бути нейтральними.
Рівняння Нернста для цієї напівреакції дорівнює
\[E = E_{\text{In}_\text{ox}/\text{In}_\text{red}}^{\circ} - \frac{0.05916}{n} \log{\frac{[\text{In}_\text{red}]}{[\text{In}_\text{ox}]}} \nonumber\]
Як показано на малюнку Template:index, якщо припустити, що колір індикатора змінюється від кольору In ox до червоного, коли співвідношення [In red]/[In ox] змінюється від 0,1 до 10, то кінцева точка виникає, коли потенціал рішення знаходиться в межах діапазону
\[E = E_{\text{In}_\text{ox}/\text{In}_\text{red}}^{\circ} \pm \frac{0.05916}{n} \nonumber\]
Це той самий підхід, який ми використовували при розгляді кислотно-базових показників та показників комплексоутворення.
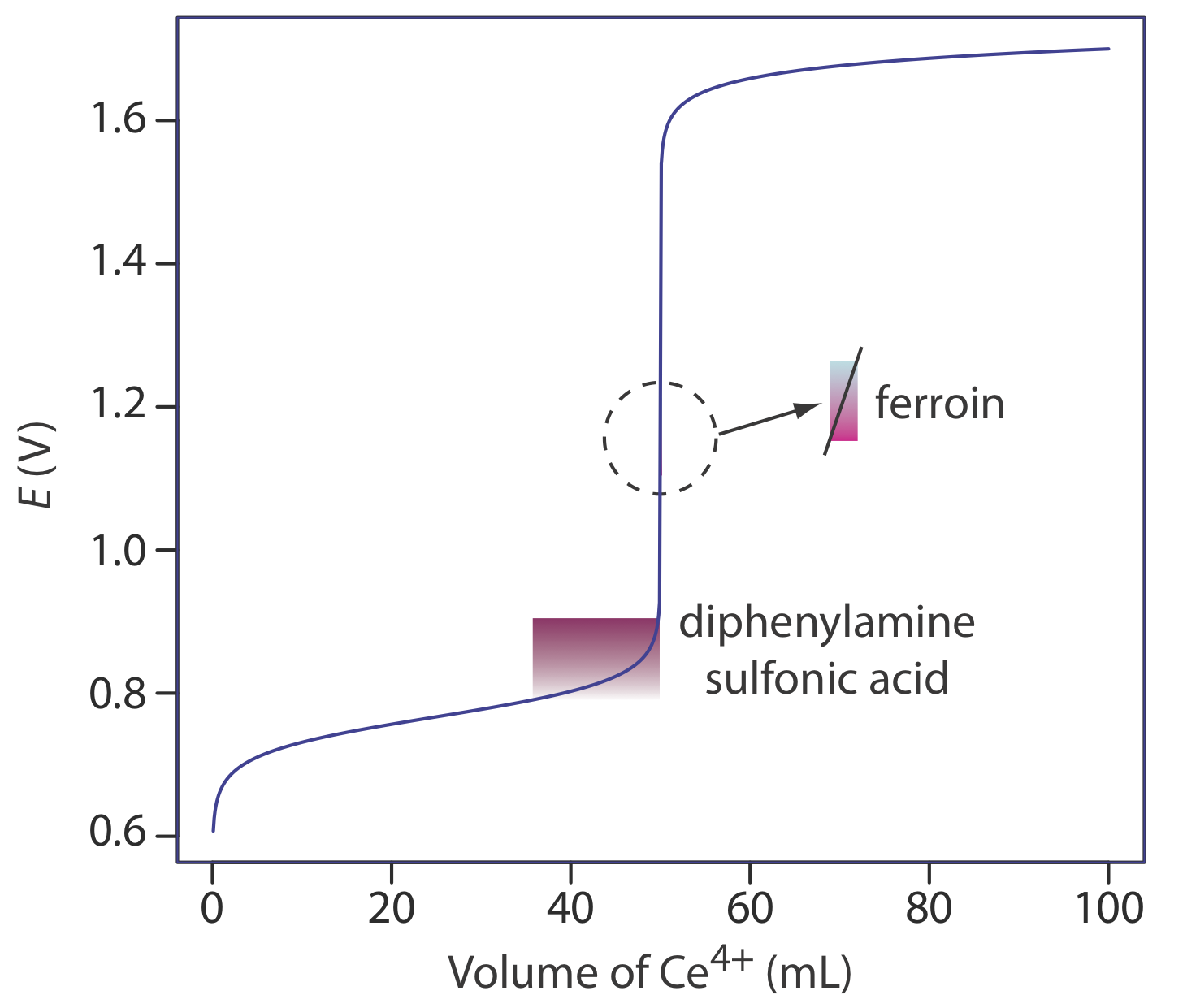
Частковий список окислювально-відновних показників наведено в таблиці Template:index. Приклади відповідного і невідповідного показника для титрування Fe 2 + з Ce 4 + наведено на малюнку Template:index.
| показник | Колір У вола | колір: Червоний | Е о (В) |
|---|---|---|---|
| індиго тетрасульфат | блакитний | безбарвна | 0,36 |
| метиленовий синій | блакитний | безбарвна | 0,53 |
| дифеніламін | фіолетовий | безбарвна | 0,75 |
| дифеніламін сульфонова кислота | червоно-фіолетовий | безбарвна | 0,85 |
| трис (2,2'-біпірадин) залізо | блідо-блакитний | червоний | 1.120 |
| фероїну | блідо-блакитний | червоний | 1.147 |
| трис (5-нітро-1,10-фенантролін) залізо | блідо-блакитний | червоно-фіолетовий | 1,25 |
Інші методи знаходження кінцевої точки
Іншим методом визначення кінцевої точки окислювально-відновного титрування є потенціометричне титрування, в якому ми відстежуємо зміну потенціалу, додаючи титрант до титранду. Кінцева точка виявляється, досліджуючи візуально криву титрування. Найпростіша експериментальна конструкція для потенціометричного титрування складається з електрода-індикатора Pt, потенціал якого регулюється окислювально-відновною напівреакцією титранта або титранта, та опорного електрода, який має фіксований потенціал. Інші методи визначення кінцевої точки титрування включають термометричне титрування та спектрофотометричне титрування.
Ви будете подальше обговорення потенціометрії в главі 11.
Представницький метод 9.4.1: Визначення загального залишкового хлору
Найкращий спосіб оцінити теоретичні та практичні деталі, розглянуті в цьому розділі, - це уважно вивчити типовий окислювально-відновно-відновний титриметричний метод. Хоча кожен метод унікальний, наступний опис визначення загального залишку хлору у воді дає повчальний приклад типової процедури. Опис тут базується на методі 4500-Cl B, опублікованому в Стандартні методи дослідження води та стічних вод, 20-е видання, Американська асоціація громадського здоров'я: Вашингтон, округ Колумбія, 1998.
Опис методу
Хлорування громадського водопостачання виробляє кілька хлорсодержащих видів, комбінована концентрація яких називається загальним залишковим хлором. Хлор присутній у різних хімічних станах, включаючи вільний залишковий хлор, який складається з Cl 2, HoCl та OCl - та комбінованого залишкового хлору, який складається з NH 2 Cl, NHCl 2 та NCl 3. Загальний залишок хлору визначається за допомогою окислювальної здатності хлору для перетворення I — в\(\text{I}_3^-\). Кількість\(\text{I}_3^-\) утвореного потім визначають шляхом титрування з Na 2 S 2 O 3 з використанням крохмалю в якості індикатора. Незалежно від його форми, загальний залишок хлору повідомляється так, ніби Cl 2 є єдиним джерелом хлору, і повідомляється як мг Cl/L.
Порядок дій
Виберіть обсяг зразка, який вимагає менше 20 мл Na 2 S 2 O 3, щоб досягти кінцевої точки. Використовуючи льодовикову оцтову кислоту, підкисліть зразок до рН між 3 і 4 і додайте близько 1 грама КІ. Титрують за допомогою Na 2 S 2 O 3 до тих пір, поки жовтий колір не\(\text{I}_3^-\) почне зникати. Додайте 1 мл розчину індикатора крохмалю і продовжуйте титрування, поки синій колір крохмалю -\(\text{I}_3^-\) комплексу не зникне (рис. Template:index). Використовуйте титрування заготовки для корекції обсягу титранту, необхідного для досягнення кінцевої точки домішок реагенту.

Питання
1. Це приклад прямого або непрямого аналізу?
Це непрямий аналіз, оскільки хлорвмісні види не реагують з титрантом. Замість цього загальний залишковий хлор окислюється I — до\(\text{I}_3^-\), а кількість його\(\text{I}_3^-\) визначають шляхом титрування з Na 2 S 2 O 3.
2. Чому процедура спирається на непрямий аналіз замість прямого титрування хлорвмісних видів, використовуючи КІ як титрант?
Оскільки загальний залишок хлору складається з шести різних видів, титрування з I - не має єдиної, чітко визначеної точки еквівалентності. При перетворенні залишкового хлору в еквівалентну кількість\(\text{I}_3^-\), непряме титрування з Na 2 S 2 O 3 має єдину корисну точку еквівалентності. Навіть якщо загальний залишок хлору є від одного виду, такого як HoCl, пряме титрування за допомогою КІ недоцільно. Оскільки продукт титрування надає жовтий колір\(\text{I}_3^-\), колір титранду буде змінюватися з кожним додаванням титранту, що ускладнює пошук відповідного показника.
3. Як окислювачі, так і відновники можуть перешкоджати цьому аналізу. Поясніть вплив кожного типу інтерференту на загальний залишок хлору.
Інтерферент, який є окислювачем, перетворює додаткові I — в\(\text{I}_3^-\). Оскільки цей додатковий\(\text{I}_3^-\) вимагає додаткового обсягу Na 2 S 2 O 3, щоб досягти кінцевої точки, ми переоцінюємо загальний залишок хлору. Якщо інтерферент є відновником, він зменшується назад до I - деякої частини, що\(\text{I}_3^-\) утворюється в результаті реакції між загальним залишковим хлором і йодидом; в результаті ми недооцінюємо загальний залишок хлору.
Кількісні програми
Хоча багато кількісних застосувань окислювально-відновної титриметрії були повторно розміщені іншими аналітичними методами, кілька важливих застосувань продовжують знаходити актуальність. У цьому розділі ми розглядаємо загальне застосування окислювально-відновлювальної титриметрії з акцентом на екологічні, фармацевтичні та промислові програми. Почнемо, однак, з короткого обговорення вибору та характеристики окислювально-відновних титрантів та методів контролю ступеня окислення титранду.
Регулювання ступеня окислення Титранда
Якщо в кількісному аналізі має використовуватися окислювально-відновне титрування, титранд спочатку повинен бути присутнім в одному ступені окислення. Наприклад, залізо визначається окислювально-відновним титруванням, при якому Ce 4 + окислює Fe 2 + до Fe 3 +. Залежно від зразка і способу підготовки проби залізо спочатку може бути присутнім як в ступені окислення +2, так і +3. Перед титруванням ми повинні зменшити будь-який Fe 3 + до Fe 2 +, якщо ми хочемо визначити загальну концентрацію заліза в зразку. Цей тип попередньої обробки здійснюється за допомогою допоміжного відновника або окислювача.
Метал, який легко окислюється - такий як Zn, Al та Ag, може служити допоміжним відновником. Метал, як намотаний дріт або порошок, додається до зразка, де він зменшує титранд. Оскільки будь-який не прореагував допоміжний відновник реагує з титрантом, його видаляють перед тим, як ми почнемо титрування, видаляючи спіральний дріт або фільтруючи.
Альтернативним методом використання допоміжного відновника є іммобілізація його в колоні. Для приготування відновної колони водну суспензію остаточно розділеного металу упаковують в скляну трубку, забезпечену пористою пробкою внизу. Зразок поміщається у верхній частині колони і переміщається по колоні під впливом сили тяжіння або вакуумного всмоктування. Довжина редукційної колони та швидкість потоку вибираються для забезпечення повного зменшення аналіта.
Використовуються дві загальні редукційні колонки. У редукторі Джонса колонка заповнена амальгамованим цинком Zn (Hg), який готують шляхом короткочасного розміщення гранул Zn в розчин HgCl 2. окислення цинку
\[\text{Zn(Hg)}(s) \rightarrow \text{Zn}^{2+}(aq) + \text{Hg}(l) + 2e^- \nonumber\]
забезпечує електрони для зниження титранду. У редукторі Вальдена колона заповнена гранульованим металом Ag. Розчин, що містить титран, підкислюється HCl і пропускається через колону, де відбувається окислення срібла.
\[\text{Ag}(s) + \text{Cl}^- (aq) \rightarrow \text{AgCl}(s) + e^- \nonumber\]
забезпечує необхідні електрони для зниження титранду. Таблиця Template:index містить зведення кількох застосувань стовпців скорочення.
| окислений титран | Вальден редуктор | Редуктор Джонса |
|---|---|---|
| Автомобіль 3 + | — | \(\text{Cr}^{3+}(aq)+e^- \rightarrow \text{Cr}^{2+}(aq)\) |
| Cu 2 + | \(\text{Cu}^{2+}(aq)+e^- \rightarrow \text{Cu}^{+}(aq)\) | \(\text{Cu}^{2+}(aq)+2e^- \rightarrow \text{Cu}(s)\) |
| Фе 3+ | \(\text{Fe}^{3+}(aq)+e^- \rightarrow \text{Fe}^{2+}(aq)\) | \(\text{Fe}^{3+}(aq)+e^- \rightarrow \text{Fe}^{2+}(aq)\) |
| ТіО 2 + | — | \(\text{TiO}^{2+} (aq) + 2\text{H}^+ (aq) + e^- \rightarrow \text{Ti}^{3+} (aq) + \text{H}_2\text{O}(l)\) |
| \(\text{MoO}_4^{2+}\) | \(\text{MoO}_2^{2+}(aq) + e^- \rightarrow \text{MoO}_2^+(aq)\) | \(\text{MoO}_2^{2+}(aq) + 4\text{H}^+(aq) + 3 e^- \rightarrow \text{Mo}^{3+}(aq) + 2\text{H}_2\text{O}(l)\) |
| \(\text{VO}_4^{+}\) | \(\text{VO}_2^{+}(aq) + 2\text{H}^+(aq) + e^- \rightarrow \text{VO}^{2+}(aq) + \text{H}_2\text{O}(l)\) | \(\text{VO}_2^{+}(aq) + 4\text{H}^+(aq) + 3e^- \rightarrow \text{V}^{2+}(aq) + 2\text{H}_2\text{O}(l)\) |
В якості допоміжних окислювачів використовуються кілька реагентів, включаючи пероксидисульфат амонію, (NH 4) 2 S 2 O 8 та перекис водню H 2 O 2. Пероксидисульфат - потужний окислювач
\[\text{S}_2\text{O}_8^{2-}(aq) + 2e^- \rightarrow 2\text{SO}_4^{2-}(aq) \nonumber\]
який здатний окислювати Mn 2 + до\(\text{MnO}_4^-\), Cr 3 + до\(\text{Cr}_2\text{O}_7^{2-}\), і Ce 3 + до Ce 4 +. Надлишок пероксидисульфату руйнується короткочасним кип'ятінням розчину. Зменшення перекису водню в кислому розчині
\[\text{H}_2\text{O}_2(aq) + 2\text{H}^+(aq) + 2e^- \rightarrow 2\text{H}_2\text{O}(l) \nonumber\]
надає ще один спосіб окислення титранду. Надлишок Н 2 О 2 руйнується короткочасним кип'ятінням розчину.
Вибір та стандартизація титранту
Якщо його використовувати кількісно, концентрація титранту повинна залишатися стабільною під час аналізу. Оскільки титрант у відновленому стані сприйнятливий до окислення повітря, більшість окислювально-відновних титрувань використовують окислювач як титрант. Існує кілька поширених окислювальних титрантів, в тому числі\(\text{MnO}_4^-\), Ce 4 +\(\text{Cr}_2\text{O}_7^{2-}\), і\(\text{I}_3^-\). Який титрант використовується часто залежить від того, наскільки легко він окислює титранд. Титранд, який є слабким відновником, потребує сильного окислювального титранту, якщо реакція титрування повинна мати відповідну кінцеву точку.
Двома найсильнішими окислювальними титрантами є\(\text{MnO}_4^-\) і Ce 4 +, для яких відновні напівреакції
\[\text{MnO}_4^-(aq) + 8\text{H}^+(aq) + 5e^- \rightleftharpoons \text{Mn}^{2+}(aq) + 4\text{H}_2\text{O}(l) \nonumber\]
\[\text{Ce}^{4+}(aq) + e^- \rightleftharpoons \text{Ce}^{3+}(aq) \nonumber\]
Розчин Ce 4 + в 1 M H 2 SO 4 зазвичай готують з первинної стандартної церієвої аміачної селітри, Ce (NO 3) 4 •2NH 4 NO 3. При приготуванні з використанням матеріалу класу реагентів, такого як Ce (OH) 4, розчин стандартизується щодо первинного стандартного відновника, такого як Na 2 C 2 O 4 або Fe 2 + (підготовлений із залізного дроту) з використанням фероїну як індикатора. Незважаючи на свою доступність як основний стандарт та простоту приготування, Ce 4 + використовується не так часто, як\(\text{MnO}_4^-\) тому, що він дорожчий.
Реакції стандартизації є
\[\text{Ce}^{4+}(aq) + \text{Fe}^{2+}(aq) \rightarrow \text{Fe}^{3+}(aq) + \text{Ce}^{3+}(aq) \nonumber\]
\[2\text{Ce}^{4+}(aq) + \text{H}_2\text{C}_2\text{O}_4(aq) \rightarrow 2\text{Ce}^{3+}(aq) + 2\text{CO}_2(g) + 2\text{H}^+(aq) \nonumber\]
Розчин\(\text{MnO}_4^-\) готують з KMnO 4, який відсутній в якості первинного стандарту. Водний розчин перманганату термодинамічно нестійкий завдяки своїй здатності окислювати воду.
\[4\text{MnO}_4^-(aq) + 2\text{H}_2\text{O}(l) \rightleftharpoons 4\text{MnO}_2(s) + 3\text{O}_2 (g) + 4\text{OH}^-(aq) \nonumber\]
Ця реакція каталізується наявністю MnO 2, Mn 2 +, теплом, світлом, наявністю кислот і підстав. Помірно стійкий розчин перманганату готують шляхом кип'ятіння його протягом години і фільтрування через спечений скляний фільтр для видалення будь-якого твердого MnO 2, що випадає в осад. Стандартизація здійснюється проти первинного стандартного відновника, такого як Na 2 C 2 O 4 або Fe 2 + (підготовлений із залізного дроту), з рожевим кольором надлишку\(\text{MnO}_4^-\) сигналізації кінцевої точки. \(\text{MnO}_4^-\)Приготований таким способом розчин стійкий протягом 1-2 тижнів, хоча слід періодично перевіряти стандартизацію.
Реакції стандартизації є
\[\text{MnO}_4^-(aq) + 5\text{Fe}^{2+}(aq) + 8\text{H}^+(aq) \rightarrow \text{Mn}^{2+}(aq) + 5\text{Fe}^{3+}(aq) + 4\text{H}_2\text{O}(l) \nonumber\]
\[2\text{MnO}_4^-(aq) + 5\text{H}_2\text{C}_2\text{O}_4(aq) + 6\text{H}^+(aq) \rightarrow 2\text{Mn}^{2+}(aq) + 10\text{CO}_2(g) + 8\text{H}_2\text{O}(l) \nonumber\]
Біхромат калію є відносно сильним окислювачем, основними перевагами якого є його доступність як основний стандарт та його довгострокова стабільність при знаходженні в розчині. Однак він не такий сильний окислювач, як\(\text{MnO}_4^-\) або Ce 4 +, що робить його менш корисним, коли титранд є слабким відновником. Його редукційна напівреакція
\[\text{Cr}_2\text{O}_7^{2-}(aq) + 14\text{H}^+(aq) + 6e^- \rightleftharpoons 2\text{Cr}^{3+}(aq) + 7\text{H}_2\text{O}(l) \nonumber\]
Хоча розчин\(\text{Cr}_2\text{O}_7^{2-}\) помаранчевий, а розчин Cr 3 + - зелений, жоден колір не є достатньо інтенсивним, щоб служити корисним показником. Дифеніламін сульфонова кислота, окислена форма якої є червоно-фіолетовою, а відновлена форма безбарвна, дає дуже виразний сигнал кінцевої точки с\(\text{Cr}_2\text{O}_7^{2-}\).
Йод - ще один важливий окислювальний титрант. Оскільки це слабший окислювач\(\text{MnO}_4^-\), ніж, Ce 4 +, і\(\text{Cr}_2\text{O}_7^{2-}\), він корисний лише тоді, коли титранд є сильнішим відновником. Це очевидне обмеження, однак, робить I 2 більш селективним титрантом для аналізу сильного відновника в присутності слабшого відновника. Редукційна напівреакція для I 2 становить
\[\text{I}_2(aq) + 2e^- \rightleftharpoons 2\text{I}^-(aq) \nonumber\]
Оскільки йод не дуже розчинний у воді, розчини готують шляхом додавання надлишку I —. Реакція комплексоутворення
\[\text{I}_2(aq) + \text{I}^-(aq) \rightleftharpoons \text{I}_3^-(aq) \nonumber\]
підвищує розчинність I 2, утворюючи більш розчинний іон трийодиду,\(\text{I}_3^-\). Незважаючи на те, що йод присутній\(\text{I}_3^-\) замість I 2, кількість електронів у відновній напівреакції не впливає.
\[\text{I}_3^-(aq) + 2e^-(aq) \rightleftharpoons 3\text{I}^-(aq) \nonumber\]
Розчини в\(\text{I}_3^-\) нормі стандартизовані проти Na 2 S 2 O 3 з використанням крохмалю в якості конкретного показника для\(\text{I}_3^-\).
Реакція стандартизації є
\[\text{I}_3^-(aq) + 2\text{S}_2\text{O}_3^{2-}(aq) \rightarrow 3\text{I}^-(aq) + 2\text{S}_4\text{O}_6^{2-} (aq) \nonumber\]
Окислювальний титрант\(\text{MnO}_4^-\), такий як, Ce 4 +\(\text{Cr}_2\text{O}_7^{2-}\)\(\text{I}_3^-\), і, використовується, коли титранд знаходиться в відновленому стані. Якщо титранд знаходиться в окисленому стані, ми можемо спочатку зменшити його допоміжним відновником, а потім завершити титрування за допомогою окислювального титранту. Крім того, ми можемо титрувати його за допомогою відновлювального титранту. Йодид є відносно сильним відновником, який може служити відновлювальним титрантом, за винятком того, що його розчини сприйнятливі до окислення повітря I - до\(\text{I}_3^-\).
\[3\text{I}^-(aq) \rightleftharpoons \text{I}_3^- (aq) + 2e^- \nonumber\]
Свіжоприготований розчин КІ прозорий, але через кілька днів він може проявити слабку жовту забарвлення через наявність\(\text{I}_3^-\).
Замість цього додавання надлишку КІ зменшує титранд і вивільняє стехіометричну кількість\(\text{I}_3^-\). Кількість\(\text{I}_3^-\) виробленого потім визначається зворотним титруванням з використанням тіосульфату\(\text{S}_2\text{O}_3^{2-}\), як відновлювального титранту.
\[2\text{S}_2\text{O}_3^{2-}(aq) \rightleftharpoons \text{S}_4\text{O}_6^{2-}(aq) + 2e^- \nonumber\]
Розчини\(\text{S}_2\text{O}_3^{2-}\) готують з використанням Na 2 S 2 O 3 •5H 2 O і стандартизують перед застосуванням. Стандартизація здійснюється шляхом розчинення ретельно зваженої частини первинного стандарту КІО 3 в кислому розчині, який містить надлишок КІ. Реакція між\(\text{IO}_3^-\) і я —
\[\text{IO}_3^-(aq) + 8\text{I}^-(aq) + 6\text{H}^+(aq) \rightarrow 3\text{I}_3^-(aq) + 3\text{H}_2\text{O}(l) \nonumber\]
звільняє стехіометричну величину I-3. Титуючи це\(\text{I}_3^-\) тіосульфатом, використовуючи крохмаль як візуальний індикатор, ми можемо визначити концентрацію\(\text{S}_2\text{O}_3^{2-}\) в титранті.
Стандартизація титрування є
\[\text{I}_3^-(aq) + 2\text{S}_2\text{O}_3^{2-}(aq) \rightarrow 3\text{I}^-(aq) + \text{S}_4\text{O}_6^{2-}(aq) \nonumber\]
яка є тією ж реакцією, яка використовується для стандартизації розчинів\(\text{I}_3^-\). Такий підхід до стандартизації розчинів аналогічний\(\text{S}_2\text{O}_2^{3-}\) тому, який використовується при визначенні загального залишкового хлору, викладеного в репрезентативному методі 9.4.1.
Хоча тіосульфат є одним з небагатьох відновлювальних титрантів, який не легко окислюється при контакті з повітрям, він схильний до повільного розкладання до бісульфіту та елементарної сірки. При використанні протягом декількох тижнів розчин тіосульфату періодично повторюється. Кілька форм бактерій здатні метаболізувати тіосульфат, що призводить до зміни його концентрації. Ця проблема зводиться до мінімуму додаванням в розчин такого консерванту, як HGi 2.
Іншим корисним відновлювальним титрантом є сульфат амонію заліза, Fe (NH 4) 2 (SO 4) 2 • 6H 2 O, в якому залізо присутнє в ступені окислення +2. Розчин Fe 2 + сприйнятливий до окислення повітря, але при приготуванні в 0,5 М Н 2 SO 4 він залишається стабільним протягом цілого місяця. Періодична рестандартизація з K 2 Cr 2 O 7 доцільна. Сульфат амонію заліза використовують як титрант при безпосередньому аналізі титранду, або, його додають в титранд в надлишку і кількість виробленого Fe 3 + визначають шляхом зворотного титрування стандартним розчином Ce 4 + або\(\text{Cr}_2\text{O}_7^{2-}\).
неорганічний аналіз
Одним з найважливіших застосувань окислювально-відновної титріметрії є оцінка хлорування громадських водопостачань. Представницький метод 9.4.1, наприклад, описує підхід до визначення загального залишку хлору за допомогою окислювальної здатності хлору для окислення I — до\(\text{I}_3^-\). Кількість\(\text{I}_3^-\) визначається шляхом зворотного титрування с\(\text{S}_2\text{O}_3^{2-}\).
Ефективність хлорування залежить від форми хлорующего виду. Існує два внески у загальний залишок хлору - залишок вільного хлору та комбінований залишковий хлор. Вільний залишок хлору включає форми хлору, які доступні для дезінфекції водопостачання. Приклади видів, які сприяють вільному залишковому хлору, включають Cl 2, HoCl та OCl -. Комбінований залишковий хлор включає ті види, у яких хлор знаходиться в відновленому вигляді і, отже, вже не здатний забезпечити дезінфекцію. Видами, які сприяють комбінованому залишковому хлору, є NH 2 Cl, NHCl 2 та NCl 3.
При змішуванні зразка безйодистої хлорованої води з перевищенням показника N, N -діетил- р -фенілендіаміну (ДПД) вільний хлор окислює стехіометричну частину ДПД до своєї червоноокрашенной форми. Окислений DPD потім назад титрують до безбарвної форми з використанням сульфату амонію заліза в якості титранту. Обсяг титранту пропорційний вільному залишковому хлору.
Визначивши залишок вільного хлору в пробі води, додають невелику кількість КІ, який каталізує відновлення монохлораміну, NH 2 Cl, і окислює частину ДПД назад до свого червоного кольору. Титрування окисленого DPD сульфатом амонію заліза дає кількість NH 2 Cl у зразку. Кількість дихлораміну і трихлораміну визначаються аналогічним чином.
Описані вище методи визначення загального, вільного або комбінованого залишкового хлору також використовуються для встановлення потреби в хлорі у водопостачанні. Потреба в хлорі визначається як кількість хлору, необхідна для повної реакції з будь-якою речовиною, яка може окислюватися хлором, зберігаючи при цьому бажаний залишок хлору. Він визначається шляхом додавання поступово більшої кількості хлору до набору зразків, взятих з водопроводу, і визначення загального, вільного або комбінованого залишкового хлору.
Ще одним важливим прикладом окислювально-відновної титриметрії, який знаходить застосування як в аналізі охорони здоров'я, так і в екологічному аналізі, є визначення розчиненого кисню. У природних водах, таких як озера та річки, рівень розчиненого O 2 важливий з двох причин: він є найбільш доступним окислювачем для біологічного окислення неорганічних та органічних забруднювачів; і він необхідний для підтримки водного життя. У очисних спорудах розчинений O 2 має важливе значення для аеробного окислення відходів. Якщо концентрація розчиненого О 2 падає нижче критичного значення, аеробні бактерії замінюються анаеробними бактеріями, а при окисленні органічних відходів утворюються небажані гази, такі як CH 4 і H 2 S.
Одним із стандартних методів визначення розчиненого O 2 в природних водах і стічних водах є метод Вінклера. Зразок води збирають, не піддаючи її атмосфері, що може змінити концентрацію розчиненого O 2. Зразок спочатку обробляють розчином MnSO 4, а потім розчином NaOH і КІ. У цих лужних умовах розчинений кисень окислює Mn 2 + до MnO 2.
\[2\text{Mn}^{2+}(aq) + 4\text{OH}^-(aq) + \text{O}_2(g) \rightarrow 2\text{MnO}_2(s) + 2\text{H}_2\text{O}(l) \nonumber\]
Після завершення реакції розчин підкислюють H 2 SO 4. У нині кислих умовах I —\(\text{I}_3^-\) окислюється до MnO 2.
\[\text{MnO}_2(s) + 3\text{I}^-(aq) + 4\text{H}^+(aq) \rightarrow \text{Mn}^{2+}(aq) + \text{I}_3^-(aq) + 2\text{H}_2\text{O}(l) \nonumber\]
Кількість цих форм\(\text{I}_3^-\) визначається титруванням з\(\text{S}_2\text{O}_3^{2-}\) використанням крохмалю як індикатора. Метод Вінклера піддається безлічі перешкод і запропоновано кілька модифікацій початкової процедури. Наприклад,\(\text{NO}_2^-\) заважає тому, що зводиться\(\text{I}_3^-\) до I — в кислих умовах. Ця перешкода усувається додаванням азиду натрію, NaN 3, який знижується\(\text{NO}_2^-\) до N 2. Інші відновники, такі як Fe 2 +, усуваються попередньою обробкою зразка KMnO 4 і знищенням надлишку перманганату K 2 C 2 O 4.
Ще одним важливим прикладом окислювально-відновної титриметрії є визначення води в неводних розчинниках. Титрант для цього аналізу відомий як реагент Карла Фішера і складається з суміші йоду, діоксиду сірки, піридину та метанолу. Оскільки концентрація піридину досить велика, I 2 і SO 2 реагують з піридином (py) з утворенням комплексів Py•I 2 і Py•SO 2. При додаванні до зразка, який містить воду, I 2 зменшується до I — і SO 2 окислюється до SO 3.
\[\text{py}\cdot\text{I}_2 + \text{py}\cdot\text{SO}_2 + \text{H}_2\text{O} + 2\text{py} \rightarrow 2\text{py}\cdot\text{HI} + \text{py}\cdot\text{SO}_3 \nonumber\]
Метанол включений для запобігання подальшої реакції Py•SO 3 з водою. Кінцева точка титрування сигналізується, коли розчин змінюється з жовтого кольору продукту на коричневий колір реагенту Карла Фішера.
Органічний аналіз
Окислювально-відновна титріметрія також використовується для аналізу органічних аналітів. Одним з важливих прикладів є визначення хімічної потреби в кисні (ХПК) природних вод і стічних вод. ХПК - це міра кількості кисню, необхідного для повного окислення всієї органічної речовини в зразку до CO 2 і H 2 O. Оскільки не робиться жодної спроби виправити органічну речовину, яка розкладається біологічно, або для повільної кінетики розкладання, ХПК завжди завищує справжня потреба зразка в кисні. Визначення ХПК особливо важливо при управлінні промисловими очисними спорудами стічних вод, де він використовується для моніторингу викиду багатих органічними відходами в комунальні каналізаційні системи або в навколишнє середовище.
ХПК зразка визначають шляхом рефлюксування його в присутності надлишку К 2 Cr 2 O 7, який служить окислювачем. Розчин підкислюють H 2 SO 4, використовуючи Ag 2 SO 4 для каталізації окислення низькомолекулярних жирних кислот. Сульфат ртуті, HgSO 4, додають в комплекс будь-який присутній хлорид, який запобігає осадженню каталізатора Ag + як AgCl. У цих умовах ефективність окислення органічної речовини становить 95— 100%. Після рефлюксу протягом двох годин розчин охолоджують до кімнатної температури і надлишок\(\text{Cr}_2\text{O}_7^{2-}\) визначають зворотним титруванням з використанням в якості титранта сульфату заліза амонію і фероїну як індикатора. Оскільки видалити повністю всі сліди органіки з реагентів складно, проводиться титрування заготовки. Різниця в кількості сульфату заліза амонію, необхідного для титрування зразка і заготовки, пропорційна ХПК.
Йод був використаний як окислювальний титрант для ряду сполук, що представляють фармацевтичний інтерес. Раніше ми відзначали, що реакція\(\text{S}_2\text{O}_3^{2-}\) з\(\text{I}_3^-\) виробляє іон тетратіонату,\(\text{S}_4\text{O}_6^{2-}\). Тетратіонатний іон насправді є димером, який складається з двох іонів тіосульфату, з'єднаних через дисульфідний (——S—) зв'язок. Таким же чином\(\text{I}_3^-\) використовується для титрування меркаптанів загальної формули RSH, утворюючи димер РССР як продукт. Амінокислоту цистеїн також можна титрувати\(\text{I}_3^-\). Продукт такого титрування - цистин, який є димером цистеїну. Трийодид також використовується для аналізу аскорбінової кислоти (вітаміну С) шляхом окислення функціональної групи ендіолу до альфа-дикетону
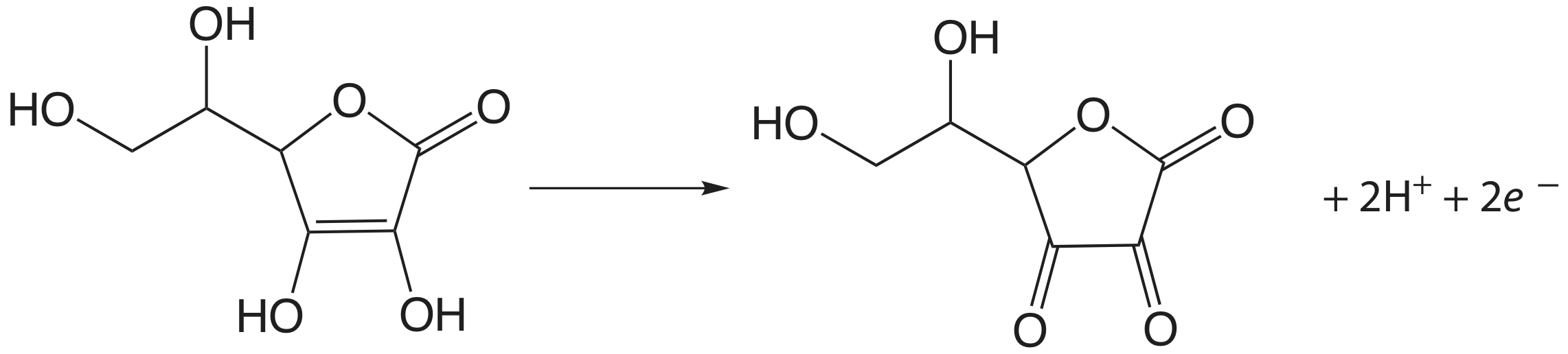
і для аналізу редукуючих цукрів, таких як глюкоза, шляхом окислення функціональної групи альдегіду до карбоксилатного іона в основному розчині.
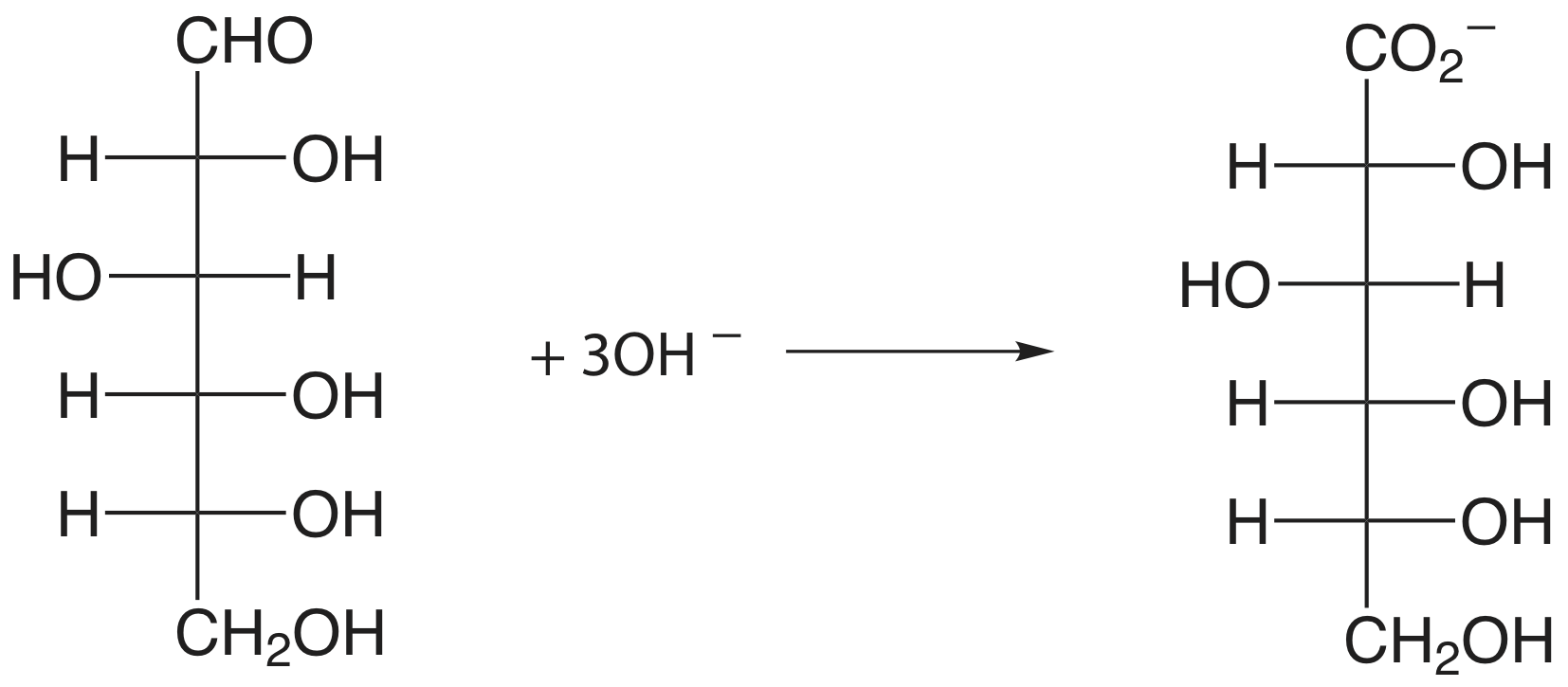
Органічна сполука, яка містить гідроксильну, карбонільну або амінну функціональну групу, прилеглу до гідроксильної або карбонільної групи, може окислюватися за допомогою метаперіодата\(\text{IO}_4^-\), як окислювального титранта.
\[\text{IO}_4^-(aq) + \text{H}_2\text{O}(l) + 2e^- \rightleftharpoons \text{IO}_3^-(aq) + 2\text{OH}^-(aq) \nonumber\]
Двоелектронне окислення розщеплює зв'язок С—С між двома функціональними групами з гідроксильними групами, окисленими до альдегідів або кетонів, карбонільними групами, окисленими до карбонових кислот, і амінами, окисленими до альдегіду та аміну (аміак, якщо первинний амін). Аналіз проводиться шляхом додавання відомого надлишку\(\text{IO}_4^-\) до розчину, який містить аналіт, і дозволяє окисленню відбуватися протягом приблизно однієї години при кімнатній температурі. Коли окислення завершиться, додається надлишок КІ, який перетворює будь-які не прореагували\(\text{IO}_4^-\) в\(\text{IO}_3^-\) і\(\text{I}_3^-\).
\[\text{IO}_4^-(aq) + 3\text{I}^-(aq) + \text{H}_2\text{O}(l) \rightarrow \text{IO}_3^-(aq) + \text{I}_3^-(aq) + 2\text{OH}^-(aq) \nonumber\]
Потім\(\text{I}_3^-\) визначається титруванням з\(\text{S}_2\text{O}_3^{2-}\) використанням крохмалю в якості індикатора.
Кількісні розрахунки
Кількісна залежність між титрандом і титрантом визначається стехіометрією реакції титрування. Якщо ви не впевнені в збалансованій реакції, ви можете вивести її стехіометрію, пам'ятаючи, що електрони в окислювально-відновній реакції зберігаються.
Кількість Fe в 0,4891-г пробі руди визначають шляхом титрування K 2 Cr 2 O 7. Після розчинення зразка в HCl залізо доводять до стану окислення +2 за допомогою редуктора Джонса. Титрування до кінцевої точки дифеніламіну сульфонової кислоти вимагає 36,92 мл 0,02153 M K 2 Cr 2 O 7. Повідомте про вміст заліза в руді як %w/w Fe 2 O 3.
Рішення
Оскільки нам не забезпечена реакція титрування, ми будемо використовувати збереження електронів для виведення стехіометрії. Під час титрування аналіт окислюється від Fe 2 + до Fe 3 +, а титрант відновлюється з\(\text{Cr}_2\text{O}_7^{2-}\) до Cr 3 +. Окислення Fe 2 + до Fe 3 + вимагає одного електрона. Зниження\(\text{Cr}_2\text{O}_7^{2-}\), при якому кожен хром знаходиться в стані окислення +6, до Cr 3+ вимагає трьох електронів на хром, в цілому шість електронів. Збереження електронів для титрування, отже, вимагає, щоб кожен моль K 2 Cr 2 O 7 реагував з шістьма молями Fe 2 +.
Родимки K 2 Cr 2 O 7, використовувані для досягнення кінцевої точки
\[(0.02153 \text{ M})(0.03692 \text{ L}) = 7.949 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7 \nonumber\]
що означає, що зразок містить
\[7.949 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7 \times \frac{6 \text{ mol Fe}^{2+}}{\text{mol K}_2\text{Cr}_2\text{O}_7} = 4.769 \times 10^{-3} \text{ mol Fe}^{2+} \nonumber\]
Таким чином, %w/w Fe 2 O 3 в зразку руди дорівнює
\[4.769 \times 10^{-3} \text{ mol Fe}^{2+} \times \frac{1 \text{ mol Fe}_2\text{O}_3}{2 \text{ mol Fe}^{2+}} \times \frac{159.69 \text{g Fe}_2\text{O}_3}{\text{mol Fe}_2\text{O}_3} = 0.3808 \text{ g Fe}_2\text{O}_3 \nonumber\]
\[\frac{0.3808 \text{ g Fe}_2\text{O}_3}{0.4891 \text{ g sample}} \times 100 = 77.86 \text{% w/w Fe}_2\text{O}_3 \nonumber\]
Хоча ми можемо вивести стехіометрію між титрантом і титрандом у прикладі Template:index без збалансування реакції титрування, збалансована реакція
\[\text{K}_2\text{Cr}_2\text{O}_7(aq) + 6\text{Fe}^{2+}(aq) + 14\text{H}^+(aq) \rightarrow 2\text{Cr}^{3+}(aq) + 2\text{K}^+(aq) + 6\text{Fe}^{3+}(aq) + 7\text{H}_2\text{O}(l) \nonumber\]
дійсно надає корисну інформацію. Наприклад, наявність Н + нагадує про те, що реакція повинна проходити в кислому розчині.
Чистота зразка оксалату натрію, Na 2 C 2 O 4, визначається титруванням стандартним розчином KMnO 4. Якщо для зразка 0,5116-г потрібно 35,62 мл 0,0400 М кМNo 4, щоб досягти кінцевої точки титрування, що є %w/w Na 2 C 2 O 4 у зразку.
- Відповідь
-
Оскільки нам не забезпечена збалансована реакція, давайте використаємо збереження електронів для виведення стехіометрії. Окислення\(\text{C}_2\text{O}_4^{2-}\), при якому кожен вуглець має ступінь окислення +3, до CO 2, в якому вуглець має ступінь окислення +4, вимагає одного електрона на вуглець або всього два електрони для кожного моля\(\text{C}_2\text{O}_4^{2-}\). Зниження\(\text{MnO}_4^-\), при якому кожен марганець знаходиться в стані окислення +7, до Mn 2 + потрібно п'ять електронів. Збереження електронів для титрування, отже, вимагає, щоб два молі KMnO 4 (10 молів е -) вступали в реакцію з п'ятьма молями Na 2 C 2 O 4 (10 молів е -).
Кроти KMnO 4, які використовуються для досягнення кінцевої точки, є
\[(0.0400 \text{ M KMnO}_4)(0.03562 \text{ L})=1.42 \times 10^{-3} \text{ mol KMnO}_4 \nonumber\]
що означає, що зразок містить
\[1 .42 \times 10^{-3} \text{ mol KMnO}_4 \times \frac{5 \text{ mol Na}_2\text{C}_2\text{O}_4}{2 \text{ mol KMnO}_4} = 3.55 \times 10^{-3} \text{ mol Na}_2\text{C}_2\text{O}_4 \nonumber\]
Таким чином, %w/w Na 2 C 2 O 4 в зразку руди дорівнює
\[3.55 \times 10^{-3} \text{ mol Na}_2\text{C}_2\text{O}_4 \times \frac{134.00 \text{ g Na}_2\text{C}_2\text{O}_4}{\text{mol Na}_2\text{C}_2\text{O}_4} = 0.476 \text{ g Na}_2\text{C}_2\text{O}_4 \nonumber\]
\[\frac{0.476 \text{ g Na}_2\text{C}_2\text{O}_4}{0.5116 \text{ g sample}} \times 100 = 93.0 \text{% w/w Na}_2\text{C}_2\text{O}_4 \nonumber\]
Як показано на наступних двох прикладах, ми можемо легко розширити цей підхід до аналізу, який вимагає непрямого аналізу або зворотного титрування.
25,00-мл проба рідкого відбілювача розводять до 1000 мл в об'ємній колбі. 25-мл частина розведеного зразка переноситься піпетом у колбу Ерленмейєра, яка містить надлишок KI, зменшуючи OCl - до Cl - і виробляє\(\text{I}_3^-\). \(\text{I}_3^-\)Вивільнений визначають титруванням 0,09892 M Na 2 S 2 O 3, що вимагає 8,96 мл для досягнення кінцевої точки показника крохмалю. Повідомте про %w/v NaOCl у зразку відбілювача.
Рішення
Щоб визначити стехіометрію між аналітом, NaOCl та титрантом Na 2 S 2 O 3, потрібно розглянути як реакцію між OCl — і I —, так і титрування\(\text{I}_3^-\) з Na 2 S 2 O 3.
По-перше, при відновленні OCl — до Cl — ступінь окислення хлору змінюється від +1 до —1, вимагаючи двох електронів. Окислення трьох I — до утворення\(\text{I}_3^-\) вивільняє два електрони, оскільки ступінь окислення кожного йоду змінюється від —1 в I - до —1⁄3 дюйма\(\text{I}_3^-\). Збереження електронів, отже, вимагає, щоб кожен моль OCl — виробляв один моль\(\text{I}_3^-\).
По-друге, в реакції титрування,\(\text{I}_3^-\) зводиться до I — і\(\text{S}_2\text{O}_3^{2-}\) окислюється до\(\text{S}_4\text{O}_6^{2-}\). Зменшення\(\text{I}_3^-\) до 3I - вимагає двох виборів, оскільки кожен йод змінюється від ступеня окислення від -1⁄3 до —1. При окисленні\(\text{S}_2\text{O}_3^{2-}\) до кожна\(\text{S}_4\text{O}_6^{2-}\) сірка змінює ступінь окислення від +2 до +2,5, виділяючи по одному електрону для кожного\(\text{S}_2\text{O}_3^{2-}\). Збереження електронів, отже, вимагає, щоб кожен моль\(\text{I}_3^-\) реагував з двома молями\(\text{S}_2\text{O}_3^{2-}\).
Нарешті, оскільки кожен моль OCl — виробляє один моль\(\text{I}_3^-\), і кожен моль\(\text{I}_3^-\) реагує з двома молями\(\text{S}_2\text{O}_3^{2-}\), ми знаємо, що кожен моль NaOCl у зразку в кінцевому підсумку призводить до споживання двох молів Na 2 S 2 O 3.
Родимки Na 2 S 2 O 3, використовувані для досягнення кінцевої точки титрування
\[(0.09892 \text{ M})(0.00896 \text{ L}) = 8.86 \times 10^{-4} \text{ mol Na}_2\text{S}_2\text{O}_3 \nonumber\]
що означає, що зразок містить
\[8.86 \times 10^{-4} \text{ mol Na}_2\text{S}_2\text{O}_3 \times \frac{1 \text{ mol NaOCl}}{\text{mol Na}_2\text{S}_2\text{O}_3} \times \frac{74.44 \text{ g NaOCl}}{\text{mol NaOCl}} = 0.03299 \text{ g NaOCl} \nonumber\]
Таким чином, %w/v NaOCl в розведеному зразку становить
\[\frac{0.03299 \text{ g NaOCl}}{25.00 \text{ mL}} \times 100 = 0.132 \text{% w/v NaOCl} \nonumber\]
Оскільки відбілювач розбавлявся коефіцієнтом\(40 \times\) (від 25 мл до 1000 мл), концентрація NaOCl у відбілювачі становить 5,28% в/в.
Збалансованими реакціями для цього аналізу є:
\[\text{OCl}^-(aq) + 3\text{I}^-(aq) + 2\text{H}^+(aq) \rightarrow \text{I}_3^-(aq) + \text{Cl}^-(aq) + \text{H}_2\text{O}(l) \nonumber\]
\[\text{I}_3^-(aq) + 2\text{S}_2\text{O}_3^{2-}(aq) \rightarrow \text{S}_4\text{O}_6^{2-}(aq) + 3\text{I}^-(aq) \nonumber\]
Кількість аскорбінової кислоти, С 6 Н 8 О 6, в апельсиновому соку визначають шляхом окислення аскорбінової кислоти до дегідроаскорбінової кислоти, С 6 Н 6 О 6, при відомій кількості\(\text{I}_3^-\), і зворотного титрування надлишку\(\text{I}_3^-\) з Na 2 S 2 АБО 3. 5,00-мл проби фільтрованого апельсинового соку обробляють 50,00 мл 0,01023 М\(\text{I}_3^-\). Після завершення окислення потрібно 13,82 мл 0,07203 M Na 2 S 2 O 3 для досягнення кінцевої точки показника крохмалю. Повідомляють про концентрацію аскорбінової кислоти в мг/100 мл.
Рішення
Для зворотного титрування нам потрібно визначити стехіометрію між\(\text{I}_3^-\) і аналітом, C 6 H 8 O 6, а між\(\text{I}_3^-\) і титрантом, Na 2 S 2 O 3. Пізніше легко, тому що ми знаємо з Прикладу Template:index, що кожен моль\(\text{I}_3^-\) реагує з двома молями Na 2 S 2 O 3.
При окисленні аскорбінової кислоти до дегідроаскорбінової кислоти ступінь окислення вуглецю змінюється від +2⁄3 в C 6 H 8 O 6 до +1 в C 6 H 6 O 6. Кожен вуглець виділяє 1⁄3 електрона, або загалом два електрони на аскорбінову кислоту. Як ми дізналися в прикладі Template:index, для зменшення\(\text{I}_3^-\) потрібно два електрони; таким чином, збереження електронів вимагає, щоб кожен моль аскорбінової кислоти споживав один моль\(\text{I}_3^-\).
Загальні молі\(\text{I}_3^-\), які реагують з C 6 H 8 O 6 і з Na 2 S 2 O 3 становить
\[(0.01023 \text{ M})(0.05000 \text{ L}) = 5.115 \times 10^{-4} \text{ mol I}_3^- \nonumber\]
Заднє титрування споживає
\[0.01382 \text{ L Na}_2\text{S}_2\text{O}_3 \times \frac{0.07203 \text{ mol Na}_2\text{S}_2\text{O}_3}{\text{ L Na}_2\text{S}_2\text{O}_3} \times \frac{1 \text{ mol I}_3^-}{2 \text{ mol Na}_2\text{S}_2\text{O}_3} = 4.977 \times 10^{-4} \text{ mol I}_3^- \nonumber\]
Віднімання родимок\(\text{I}_3^-\), які реагують з Na 2 S 2 O 3 із загальної кількості родимок,\(\text{I}_3^-\) дає родимки, що реагують з аскорбіновою кислотою.
\[5.115 \times 10^{-4} \text{ mol I}_3^- - 4.977 \times 10^{-4} \text{ mol I}_3^- = 1.38 \times 10^{-5} \text{ mol I}_3^- \nonumber\]
Грами аскорбінової кислоти в 5,00-мл зразка апельсинового соку становлять
\[1.38 \times 10^{-5} \text{ mol I}_3^- \times \frac{1 \text{ mol C}_6\text{H}_8\text{O}_6}{\text{mol I}_3^-} \times \frac{176.12 \text{ g C}_6\text{H}_8\text{O}_6}{\text{mol C}_6\text{H}_8\text{O}_6} = 2.43 \times 10^{-3} \text{ g C}_6\text{H}_8\text{O}_6 \nonumber\]
У зразку 5,00 мл аскорбінової кислоти є 2,43 мг, або 48,6 мг на 100 мл апельсинового соку.
Збалансованими реакціями для цього аналізу є:
\[\text{C}_6\text{H}_8\text{O}_6(aq) + \text{I}_3^- (aq) \rightarrow 3\text{I}^-(aq) + \text{C}_6\text{H}_6\text{O}_6(aq) + 2\text{H}^+(aq) \nonumber\]
\[\text{I}_3^-(aq) + 2\text{S}_2\text{O}_3^{2-}(aq) \rightarrow \text{S}_4\text{O}_6^{2-}(aq) + 3\text{I}^-(aq) \nonumber\]
Кількісний аналіз на етанол, C 2 H 6 O, здійснюється шляхом окисно-відновного зворотного титрування. Етанол окислюється до оцтової кислоти, C 2 H 4 O 2, використовуючи надлишок дихромату\(\text{Cr}_2\text{O}_7^{2-}\), який знижується до Cr 3 +. Надлишок дихромату титрують Fe 2 +, даючи Cr 3 + та Fe 3 + як продукти. При типовому аналізі 5,00-мл проба коньяку розводять до 500 мл в об'ємній колбі. Береться проба 10,00 мл і шляхом дистиляції видаляють етанол і збирають в 50,00 мл підкисленого розчину 0,0200 М К 2 Cr 2 O7. Для зворотного титрування не відреагували\(\text{Cr}_2\text{O}_7^{2-}\) потрібно 21,48 мл 0,1014 M Fe 2 +. Обчисліть% w/v етанолу в коньяку.
- Відповідь
-
Для зворотного титрування нам потрібно визначити стехіометрію між\(\text{Cr}_2\text{O}_7^{2-}\) і аналітом, C 2 H 6 O, а між\(\text{Cr}_2\text{O}_7^{2-}\) і титрантом, Fe 2 +. При окисленні етанолу до оцтової кислоти ступінь окислення вуглецю змінюється від -2 в C 2 H 6 O до 0 в C 2 H 4 O 2. Кожен вуглець виділяє два електрони, або в цілому чотири електрони на C 2 H 6 O. При відновленні\(\text{Cr}_2\text{O}_7^{2-}\), при якому кожен хром має ступінь окислення +6, до Cr 3 +, кожен хром втрачає три електрони, в цілому шість електронів на\(\text{Cr}_2\text{O}_7^{2-}\). Для окислення Fe 2 + до Fe 3 + потрібен один електрон. Збереження електронів вимагає, щоб кожен моль K 2 Cr 2 O 7 (6 молів e —) реагував з шістьма молями Fe 2 + (6 молів e —), і що чотири молі K 2 Cr 2 О 7 (24 молі е —) реагують з шістьма молями С 2 Н 6 О (24 молі е —).
Сумарні молі К 2 Ср 2 О 7, які вступають в реакцію з С 2 Н 6 О і з Fe 2 + становить
\[(0.0200 \text{ M K}_2\text{Cr}_2\text{O}_7)(0.05000 \text{ L})=1.00 \times 10^{-3} \text{ mol K}_2\text{Cr}_2\text{O}_7 \nonumber\]
Заднє титрування з Fe 2 + споживає
\[(0.1014 \text{ M Fe}^{2+})(0.02148 \text{ L}) \times \frac{1 \text{ mol K}_2\text{Cr}_2\text{O}_7}{6 \text{ mol Fe}^{2+}} = 3.63 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7 \nonumber\]
Віднімання родимок K 2 Cr 2 O 7, які реагують з Fe 2 + із сумарних родимок K 2 Cr 2 O 7, дає родимки, які вступають в реакцію з аналітом.
\[(1.00 \times 10^{-3} \text{ mol K}_2\text{Cr}_2\text{O}_7) - (3.63 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7) = 6.37 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7 \nonumber\]
Грами етанолу в пробі 10,00-мл розведеного коньяку становлять
\[6.37 \times 10^{-4} \text{ mol K}_2\text{Cr}_2\text{O}_7 \times \frac{6 \text{ mol C}_2\text{H}_6\text{O}}{4 \text{ mol K}_2\text{Cr}_2\text{O}_7} \times \frac{46.07 \text{ g C}_2\text{H}_6\text{O}}{\text{mol C}_2\text{H}_6\text{O}} = 0.0440 \text{ g C}_2\text{H}_6\text{O} \nonumber\]
% w/v C 2 H 6 O в коньяку становить
\[\frac{0.0440 \text{ g C}_2\text{H}_6\text{O}}{10.0 \text{ mL diluted brandy}} \times \frac{500.0 \text{ mL diluted brandy}}{5.00 \text{ mL brandy}} \times 100 = 44.0 \text{% w/v C}_2\text{H}_6\text{O} \nonumber\]
Оцінка окислювально-відновної титріметрії
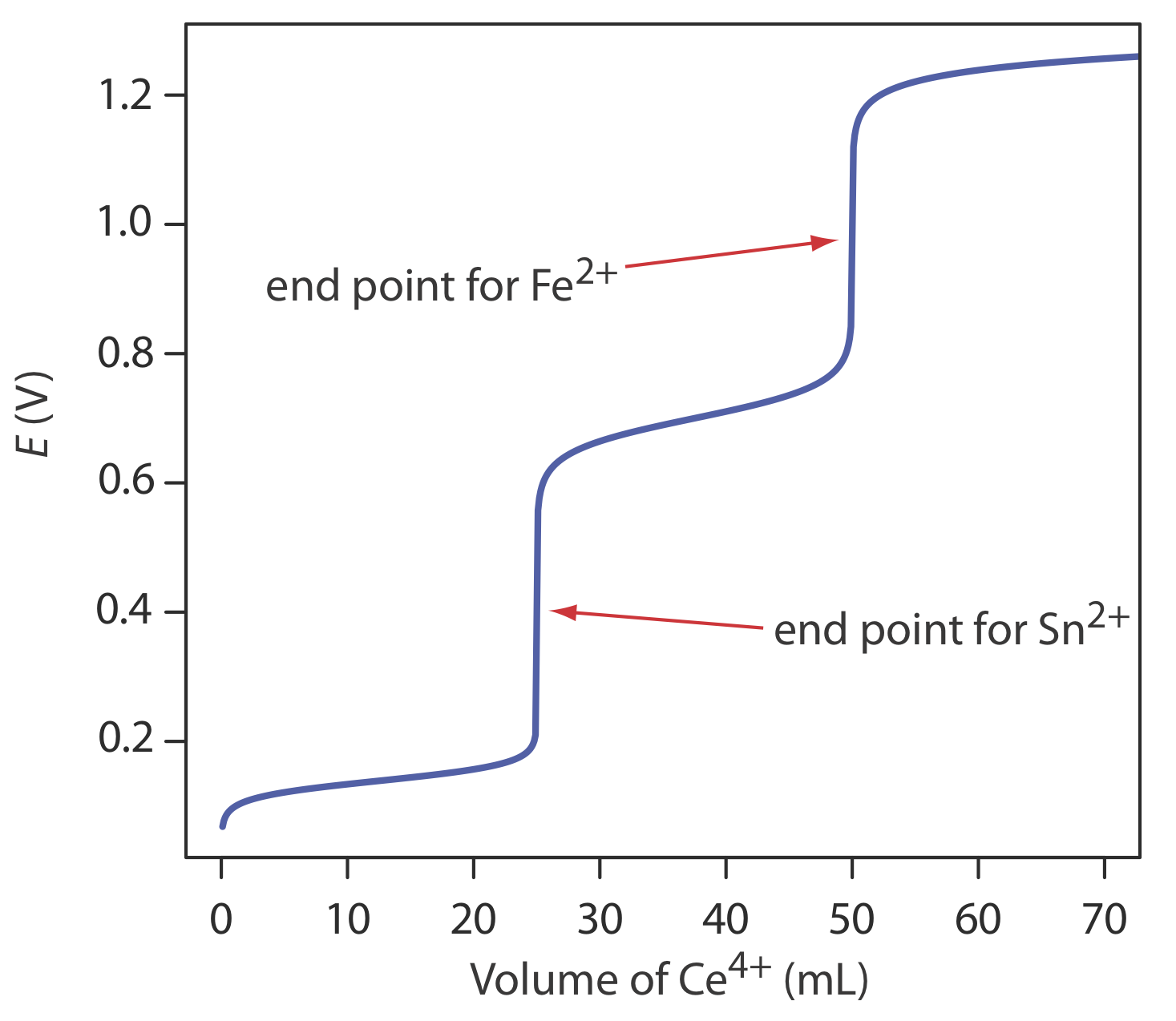
Шкала операцій, точність, точність, чутливість, час та вартість окислювально-відновного титрування подібні до описаних раніше в цьому розділі для титрування кислотно-основи або комплексоутворення. Як і при кислотно-лужному титруванні, ми можемо поширити окислювально-відновне титрування на аналіз суміші аналітів, якщо є значна різниця в їх потенціалах окислення або відновлення. На малюнку Template:index показано приклад кривої титрування для суміші Fe 2 + та Sn 2 + з використанням Ce 4 + як титранта. Титрування суміші аналітів можливо, якщо їх стандартні потенціали стану або формальні потенціали відрізняються не менше ніж на 200 мВ.