28.4: Перегородна хроматографія
- Page ID
- 27313

З безлічі форм рідинної хроматографії найбільш поширена перегородкова хроматографія. У перегородковій хроматографії час утримання розчиненої речовини визначається тим, наскільки він рухається з рухомої фази в стаціонарну фазу, а від стаціонарної фази назад в рухливу фазу. Ступінь цього рівноважного поділу визначається полярністю розчинених речовин, стаціонарної фази та рухомої фази. У хроматографії з нормальною фазою, стаціонарна фаза є полярною, а рухома фаза неполярна (або низької полярності), причому більша кількість полярних розчинів займає більше часу, щоб елютувати, оскільки вони сильніше утримуються полярною стаціонарною фазою. У хроматографії зворотної фази перегородки стаціонарна фаза неполярна, а рухлива фаза - полярна, причому більше полярних розчинів елююють швидше, оскільки вони менш сильно утримуються стаціонарною фазою. З двох режимів більш поширена зворотно-фазова перегородкова хроматографія.
Стаціонарні фази для розділової хроматографії
У перегородковій хроматографії стаціонарна фаза являє собою рідку плівку, нанесену на пакувальний матеріал, як правило, 3-10 мкм пористих частинок кремнезему. Оскільки стаціонарна фаза може бути частково розчинна в рухомій фазі, вона може елютувати або кровоточити з колони з часом. Щоб запобігти втраті стаціонарної фази, яка скорочує термін служби колони, вона ковалентно пов'язана з частинками кремнезему. Зв'язані стаціонарні фази створюються шляхом взаємодії частинок кремнезему з органохлорсиланом загальної форми Si (CH 3) 2 rCl, де R - алкільна або заміщена алкільна група.
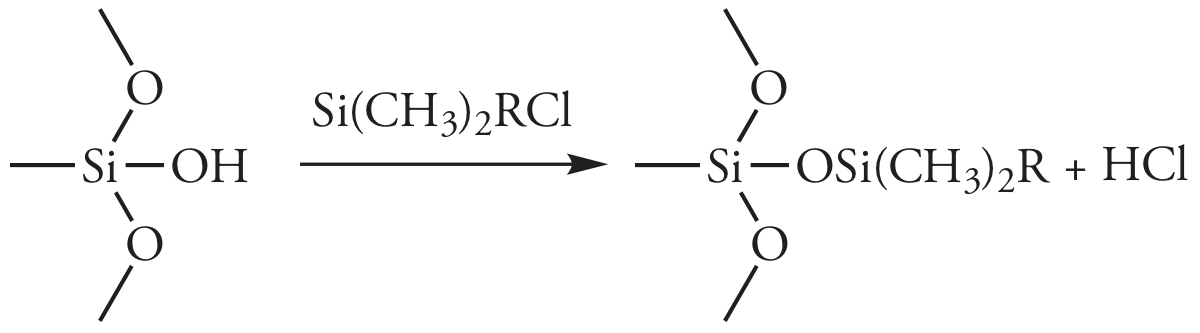
Щоб запобігти небажаним взаємодіям між розчиненими речовинами та будь-якими іншими групами —SiOH, Si (CH 3) 3 Cl використовується для перетворення сайтів, які не реагували\(–\text{SiOSi(CH}_3)_3\); такі стовпці позначаються як кінцеві.
Властивості стаціонарної фази залежать від алкільної групи органосилану. Якщо R - полярна функціональна група, то стаціонарна фаза - полярна. Приклади полярних стаціонарних фаз включають ті, де R містить ціано (—C 2 H 4 CN), діол (—C 3 H 6 OCH 2 CHOHCH 2 OH) або аміногенну (—C 3 H 6 NH 2) функціональну групу. Найбільш поширені неполярні стаціонарні фази використовують органохлорсилан, де група R - це вуглеводневий ланцюг n -октил (C 8) або n -октилдецил (С 18). Більшість розділень зворотної фази здійснюються з використанням буферного водного розчину як полярної рухомої фази або з використанням інших полярних розчинників, таких як метанол та ацетонітрил. Оскільки кремнеземний субстрат може піддаватися гідролізу в основних розчині, рН рухомої фази повинен бути менше 7,5.
Здається дивним, що більш поширена форма рідинної хроматографії ідентифікується як зворотна фаза замість нормальної фази. Одним з найбільш ранніх прикладів хроматографії було поділ Михайла Цветта рослинних пігментів, який використовував полярний стовп карбонату кальцію і неполярну рухливу фазу петролейного ефіру. Отже, присвоєння нормального та зворотного значення - це все про пріоритет.
Мобільні фази для перегородкової хроматографії
Порядок елюції розчинених речовин у ВЕРХ регулюється полярністю. Для нормального поділу фаз розчинена речовина нижчої полярності витрачає пропорційно менше часу в полярній стаціонарній фазі і елюює перед більш полярним розчиненою речовиною. Враховуючи певну стаціонарну фазу, час утримання в нормальній фазі ВЕРХ контролюється регулюванням властивостей рухомої фази. Наприклад, якщо роздільна здатність між двома розчиненими речовинами бідна, перехід на менш полярну рухливу фазу зберігає розчинені речовини на колонці протягом більш тривалого часу і надає більше можливостей для їх поділу. У ВЕРХ із зворотною фазою порядок елюції протилежний, ніж при розділенні нормальної фази, при першому елююванні більшої кількості полярних розчинів. Збільшення полярності рухомої фази призводить до більш тривалого часу утримання. Більш короткі терміни утримання вимагають рухливої фази нижчої полярності.
Вибір мобільної фази: використання індексу полярності
Існує кілька індексів, які допомагають у виборі рухомої фази, одним з яких є індекс полярності [Снайдер, Л.Р.; Glajch, J. L.; Kirkland, J. Практична розробка методу ВЕРХ, Wiley-Inter- наука: Нью-Йорк, 1988]. Таблиця Template:index містить значення індексу полярності\(P^{\prime}\), для декількох поширених рухомих фаз, де більші значення\(P^{\prime}\) відповідають більшій кількості полярних розчинників. Змішування двох або більше мобільних фаз, припускаючи, що вони змішуються - створює рухливу фазу проміжної полярності. Наприклад, бінарна рухлива фаза, зроблена шляхом поєднання розчинника А і розчинника В, має індекс полярності\(P_{AB}^{\prime}\),
\[P_{A B}^{\prime}=\Phi_{A} P_{A}^{\prime}+\Phi_{B} P_{B}^{\prime} \label{12.1} \]
де\(P_A^{\prime}\) і\(P_B^{\prime}\) є показниками полярності для розчинників A і B,\(\Phi_A\) а\(\Phi_B\) також об'ємні частки для двох розчинників.
| рухлива фаза | індекс полярності (\(P^{\prime}\)) | УФ-відсічення (нм) |
|---|---|---|
| циклогексан | \ (P^ {\ прайм}\)) ">0.04 | 210 |
| n -гексан | \ (P^ {\ прайм}\)) ">0.1 | 210 |
| чотирихлористий вуглець | \ (P^ {\ прайм}\)) ">1.6 | 265 |
| i-пропіловий ефір | \ (P^ {\ прайм}\)) ">2.4 | 220 |
| толуолу | \ (P^ {\ прайм}\)) ">2.4 | 286 |
| діетиловий ефір | \ (P^ {\ прайм}\)) ">2.8 | 218 |
| тетрагідрофуран | \ (P^ {\ прайм}\)) ">4.0 | 220 |
| етанолу | \ (P^ {\ прайм}\)) ">4.3 | 210 |
| етилацетат | \ (P^ {\ прайм}\)) ">4.4 | 255 |
| діоксан | \ (P^ {\ прайм}\)) ">4.8 | 215 |
| метанол | \ (P^ {\ прайм}\)) ">5.1 | 210 |
| ацетонітрил | \ (P^ {\ прайм}\)) ">5.8 | 190 |
| вода | \ (P^ {\ прайм}\)) ">10.2 | — |
Розділення ВЕРХ зворотної фази здійснюється за допомогою рухомої фази 60% в/в води і 40% в/в метанолу. Що таке індекс полярності рухомої фази?
Рішення
Використовуючи рівняння\ ref {12.1} та значення в таблиці Template:index, індекс полярності суміші вода-метанол 60:40 дорівнює
\[P_{A B}^{\prime}=\Phi_\text{water} P_\text{water}^{\prime}+\Phi_\text{methanol} P_\text{methanol}^{\prime} \nonumber \]
\[P_{A B}^{\prime}=0.60 \times 10.2+0.40 \times 5.1=8.2 \nonumber \]
Припустимо, вам потрібна рухлива фаза з показником полярності 7,5. Поясніть, як можна підготувати цю рухливу фазу за допомогою метанолу та води.
- Відповідь
-
Якщо ми дозволимо х бути часткою води в рухомій фазі, то 1 — х - частка метанолу. Підставляємо ці значення в рівняння\ ref {12.1} та розв'язуємо для x
\[7.5=10.2 x+5.1(1-x) \nonumber \]
\[7.5=10.2 x+5.1-5.1 x \nonumber \]
\[2.4=5.1 x \nonumber \]
дає х як 0,47. Рухливою фазою є 47% в/в води і 53% в/в метанолу.
Як правило, двоодинична зміна індексу полярності відповідає приблизно 10-кратній зміні коефіцієнта утримання розчиненої речовини. Ось простий приклад. Якщо коефіцієнт утримання розчиненої речовини, k, становить 22 при використанні води в якості рухомої фази (\(P^{\prime}\)= 10,2), то перехід на рухливу фазу 60:40 вода-метанол (\(P^{\prime}\)= 8,2) зменшується до приблизно 2,2. Зверніть увагу, що коефіцієнт утримання стає меншим, оскільки ми переходимо від більш полярної рухомої фази до менш полярної рухомої фази при розділенні зворотної фази.
Вибір мобільної фази: регулювання селективності
Зміна індексу полярності мобільної фази змінює коефіцієнт утримання розчиненої речовини. Однак, як ми дізналися в розділі 26.4, зміна k не є ефективним способом покращення роздільної здатності, коли початкове значення k перевищує 10. Для кращого поділу між двома розчиненими речовинами ми повинні покращити коефіцієнт селективності\(\alpha\). Існує два загальних методу збільшення\(\alpha\): додавання в рухому фазу реагенту, який реагує з розчиненими речовинами в реакції вторинної рівноваги або перехід на іншу рухливу фазу.
Скористатися реакцією вторинної рівноваги є корисною стратегією для поліпшення поділу [(a) Фолі, J.P. Хроматографія, 1987, 7, 118—128; (б) Фолі, J.P.; May, W.E. Anal. Хім. 1987, 59, 102—109; (с) Фолі, Дж. П.; Мей, В. Е. Хім. 1987, 59, 110—115]. На малюнку Template:index показано розділення чотирьох слабких кислот у зворотній фазі - бензойної кислоти, терефталевої кислоти, p -амінобензойної кислоти та р-гідроксибензойної кислоти - на неполярній колонці C 18, використовуючи водний буфер оцтової кислоти та ацетату натрію як рухому фазу. Час утримання цих слабких кислот коротший при використанні менш кислої рухомої фази, оскільки кожна розчинена речовина присутня в аніонній, слабкій формі основи, яка менш розчинна в неполярній стаціонарній фазі. Якщо рН рухливої фази досить кислий, розчинені речовини присутні у вигляді нейтральних слабких кислот, які більш розчинні в стаціонарній фазі і займають більше часу для елюції. Оскільки слабкі розчинені речовини кислоти не мають однакових значень p K a, рН рухомої фази по-різному впливає на час утримання кожного розчиненого речовини, що дозволяє нам знайти оптимальний рН для повного поділу чотирьох розчинених речовин.
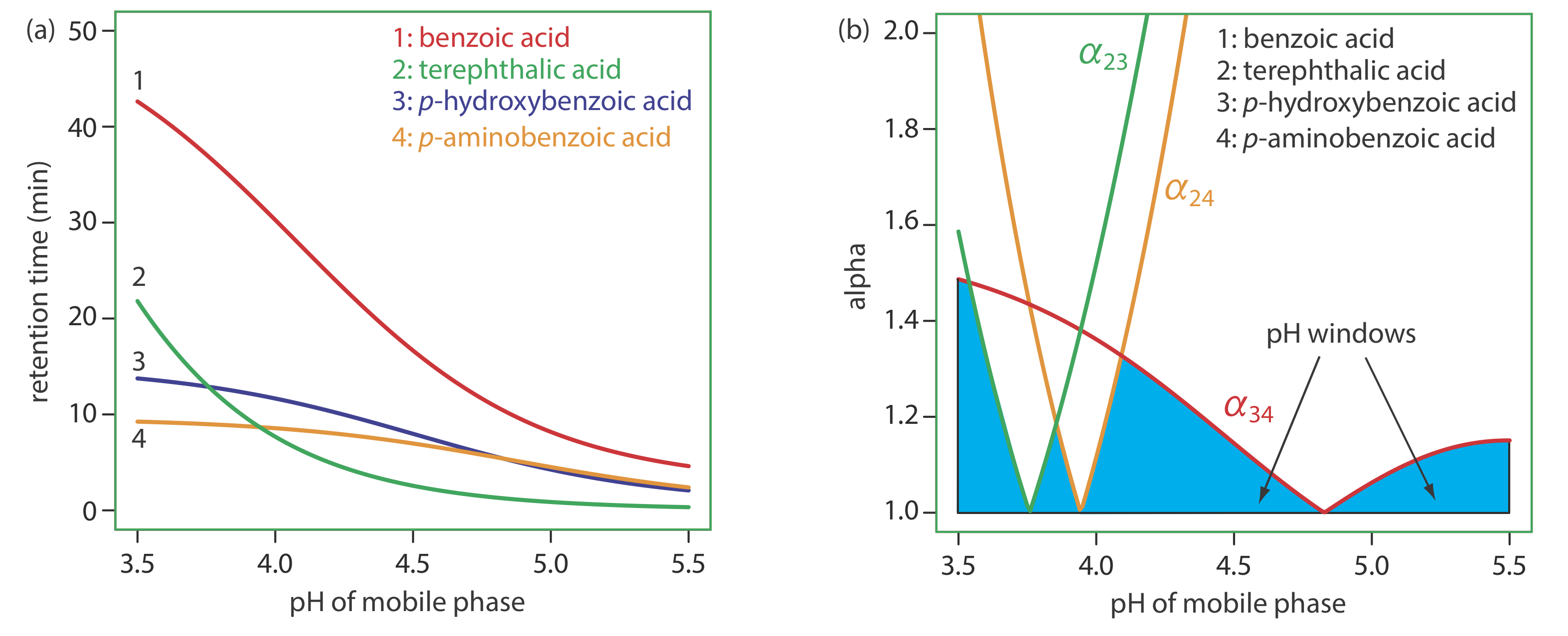
У прикладі Template:index ми навчилися регулювати полярність мобільної фази шляхом змішування двох розчинників. Індекс полярності, однак, є лише орієнтиром, і бінарні рухливі фазові суміші з однаковими показниками полярності можуть не однаково вирішити пару розчинених речовин. Наприклад, таблиця Template:index} показує час утримання чотирьох слабких кислот у двох рухомих фазах з майже однаковими значеннями для\(P^{\prime}\). Хоча порядок елюції однаковий для обох рухомих фаз, на час утримання кожного розчиненого речовини по-різному впливає вибір органічного розчинника. Якщо ми перейдемо від використання ацетонітрилу до тетрагідрофурану, наприклад, ми виявимо, що бензойна кислота елююється швидше і що р -гідроксибензойна кислота елююється повільніше. Хоча ми можемо повністю вирішити ці дві розчинні речовини, використовуючи рухливу фазу, яка становить 16% в/в ацетонітрилу, ми не можемо їх вирішити, якщо рухлива фаза становить 10% тетрагідрофурану.
| час утримання (хв) |
16% ацетонітрилу (СН 3 CN) 84% рН 4,11 водний буфер (\(P^{\prime}\)= 9,5) |
10% тетрагідрофуран (THF) 90% рН 4,11 водний буфер (\(P^{\prime}\)= 9,6) |
|---|---|---|
| \(t_\text{r, BA}\) | \ (P^ {\ прайм}\) = 9.5) ">5.18 | \ (P^ {\ прайм}\) = 9.6) ">4.01 |
| \(t_\text{r, PH}\) | \ (P^ {\ прайм}\) = 9.5) ">1.67 | \ (P^ {\ прайм}\) = 9.6) ">2.91 |
| \(t_\text{r, PA}\) | \ (P^ {\ прайм}\) = 9.5) ">1.21 | \ (P^ {\ прайм}\) = 9.6) ">1.05 |
| \(t_\text{r, TP}\) | \ (P^ {\ прайм}\) = 9.5) ">0.23 | \ (P^ {\ прайм}\) = 9.6) ">0.54 |
| \ (Р^ {\ прайм}\) = 9.6)» клас = "lt-chem-362587">
Ключ: BA - бензойна кислота; РН - р -гідроксибензойна кислота; ПА - р -амінобензойна кислота; TP - терефталева кислота |
||
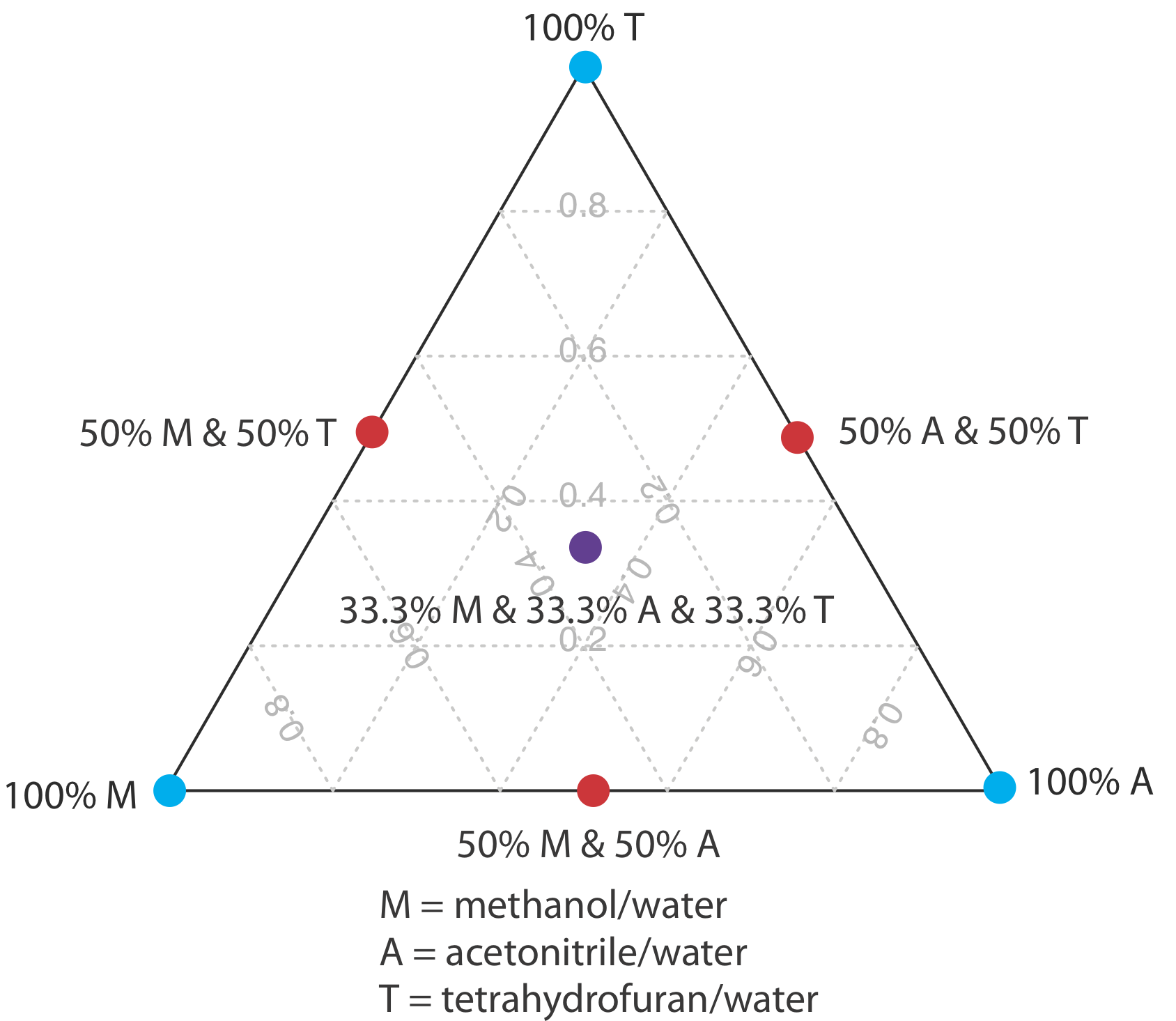
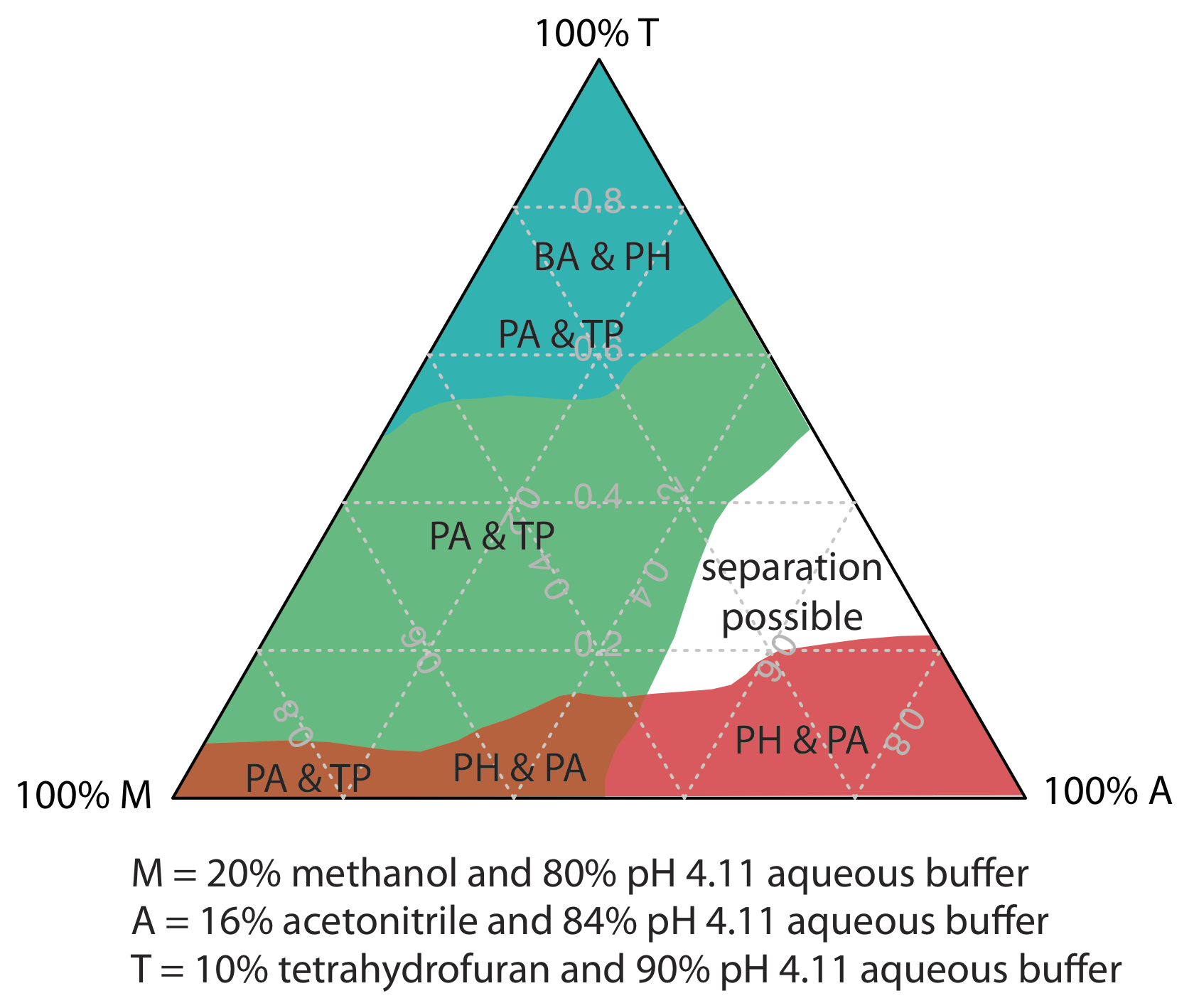
Однією зі стратегій пошуку найкращої мобільної фази є використання трикутника розчинника, показаного на рисунку Template:index, що дозволяє досліджувати широкий спектр мобільних фаз лише за сім експериментів. Ми починаємо з регулювання кількості ацетонітрилу в рухомій фазі для отримання найкращого можливого поділу протягом бажаного часу аналізу. Далі ми використовуємо таблицю Template:index для оцінки складу рухливих фаз метанолу/H 2 O та тетрагідрофуран/H 2 O, які дадуть аналогічний час аналізу. Чотири додаткові мобільні фази готуються за допомогою бінарної та потрійної рухомих фаз, показаних на рисунку Template:index. Коли ми вивчаємо хроматограми з цих семи мобільних фаз, ми можемо виявити, що одна або кілька забезпечує адекватне поділ, або ми можемо визначити область всередині трикутника розчинника, де можливе поділ. Рисунок Template:index показує карту роздільної здатності для зворотної фази поділу бензойної кислоти, терефталевої кислоти, p -амінобензойної кислоти та p -гідроксибензойної кислоти на неполярній колонці C 18, в якій максимальний бажаний час аналізу встановлено 6 хв [Harvey, D.T.; Байерлі, С.; Боумен, А.; Томлін, Дж. Чем. Едук. 1991, 68, 162—168]. Області синього, зеленого та червоного кольорів показують рухомі фазові композиції, які не забезпечують базову роздільну здатність. Незатінена область являє собою рухомі фазові композиції, де можливе поділ.
Вибір для початку з ацетонітрилу є довільним - ми можемо так само легко вибрати, щоб почати з метанолу або з тетрагідрофураном.
| % в/в СН 3 ОН | % в/в СН 3 СН | % в/в ТГФ |
|---|---|---|
| 0 | 0 | 0 |
| 10 | 6 | 4 |
| 20 | 14 | 10 |
| 30 | 22 | 16 |
| 40 | 32 | 24 |
| 50 | 40 | 30 |
| 6 | 50 | 36 |
| 70 | 60 | 44 |
| 80 | 72 | 52 |
| 90 | 87 | 62 |
| 100 | 99 | 71 |
Вибір мобільної фази: ізократичні та градієнтні елюції
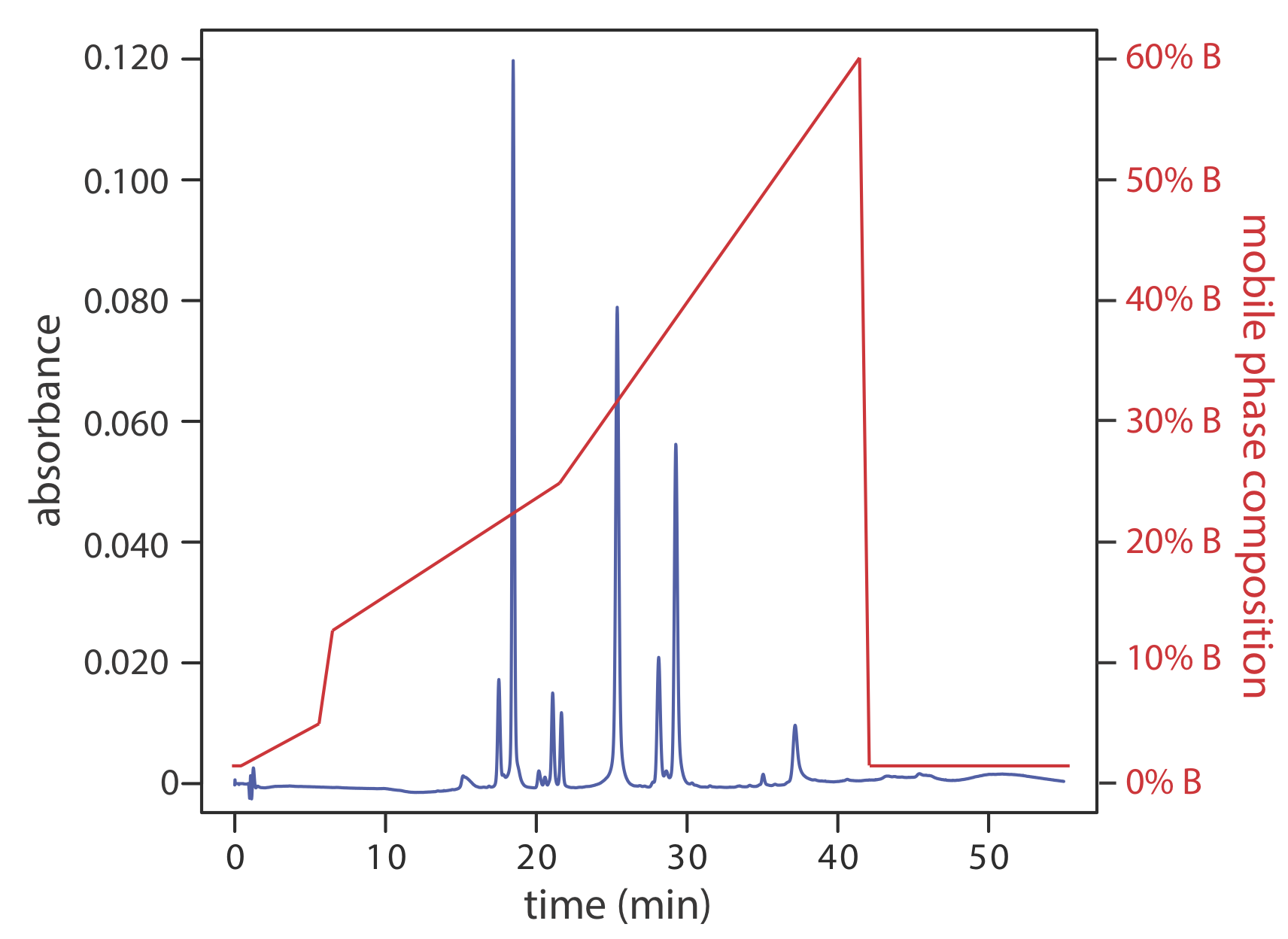
Поділ за допомогою рухливої фази, що має фіксований склад, - це і в сократичному елюції. Однією з труднощів з ізкратичним елююванням є те, що відповідна міцність рухомої фази для розчинення ранніх елюючих розчинів може призвести до неприпустимо тривалого часу утримання розчинених речовин, що запізнилися. З іншого боку, оптимізація рухомої фази для розчинів з пізнім елююванням може забезпечити недостатнє поділ розчинених речовин, що рано елютують. Зміна складу рухомої фази в міру прогресування поділу є одним з рішень цієї проблеми. Для розділення зворотної фази ми використовуємо початкову рухливу фазу, яка є більш полярною. У міру поділу ми коригуємо склад рухомої фази так, щоб вона стала менш полярною (див. Рисунок Template:index). Такі поділи називаються градієнтними елюціями.
Вибір детектора
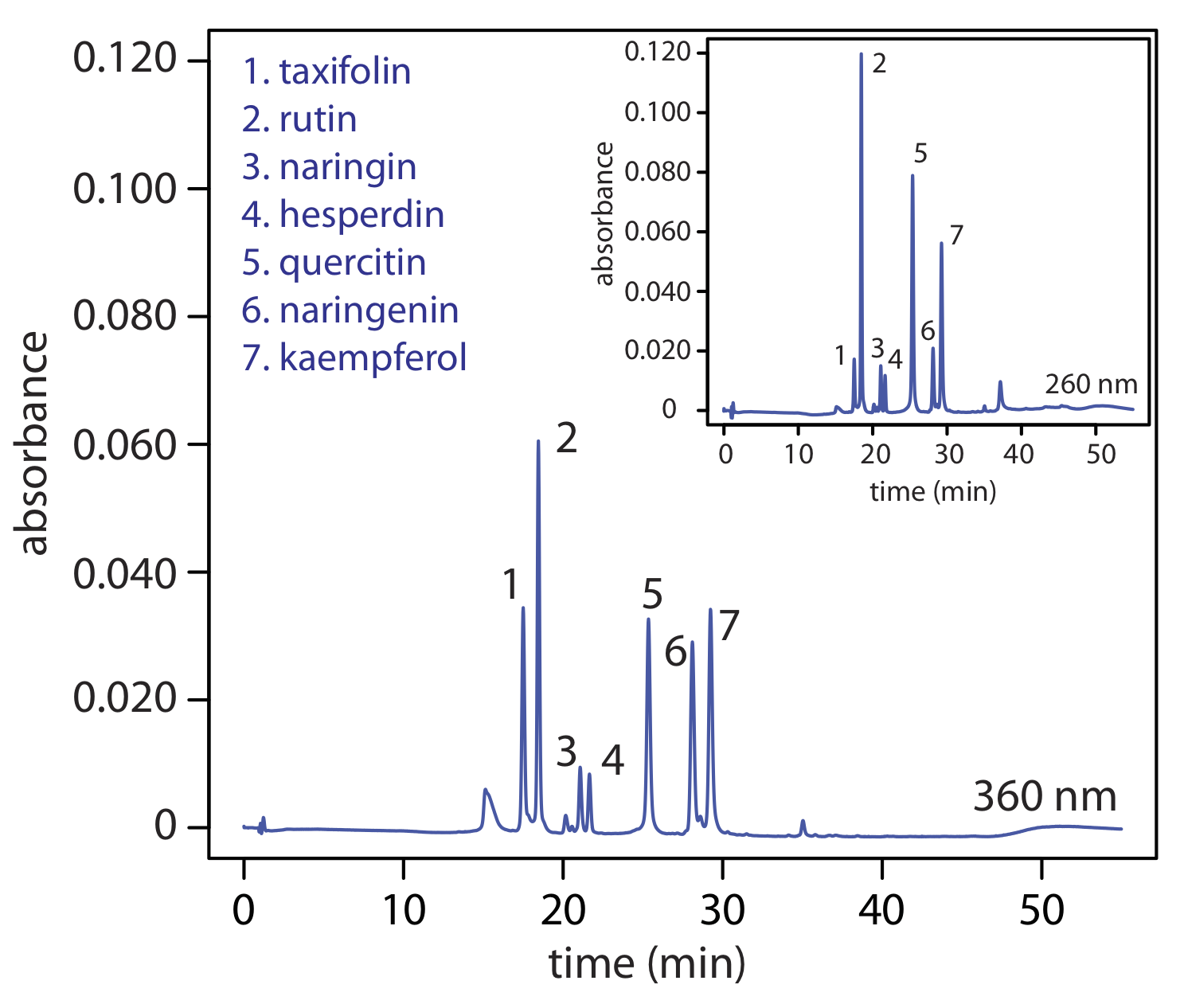
Наявність різних типів детекторів забезпечує ще один спосіб побудови селективності в аналіз. Наприклад, на малюнку Template:index} показано зворотно-фазове поділ суміші флавоноїдів за допомогою виявлення UV/Vis на двох різних довжині хвиль. У цьому випадку довжина хвилі 260 нм збільшує чутливість методу до рутину відносно таксифоліну.
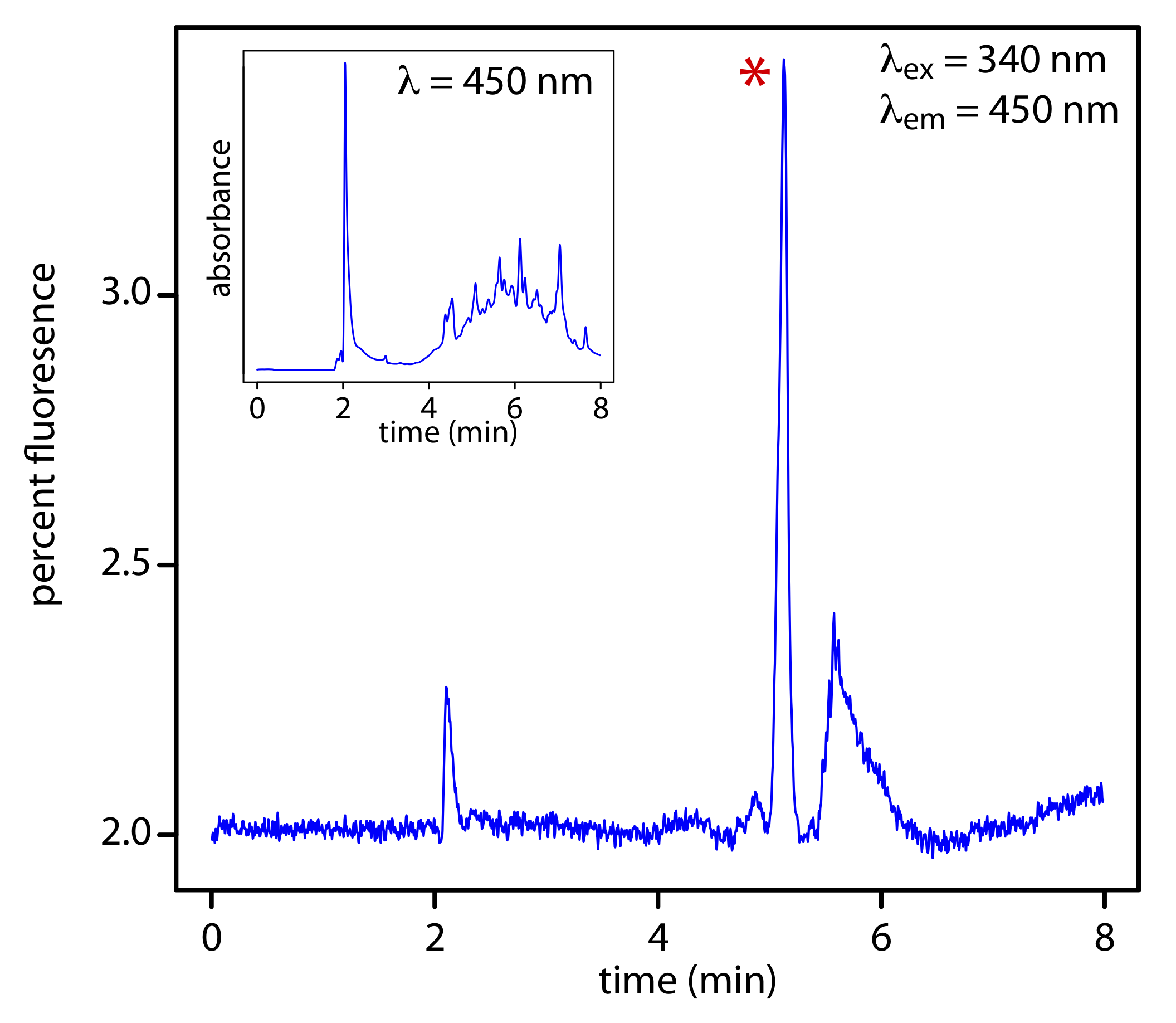
Як показано на малюнку Template:index, флуоресцентний детектор забезпечує додаткову селективність, оскільки лише деякі компоненти зразка є флуоресцентними.
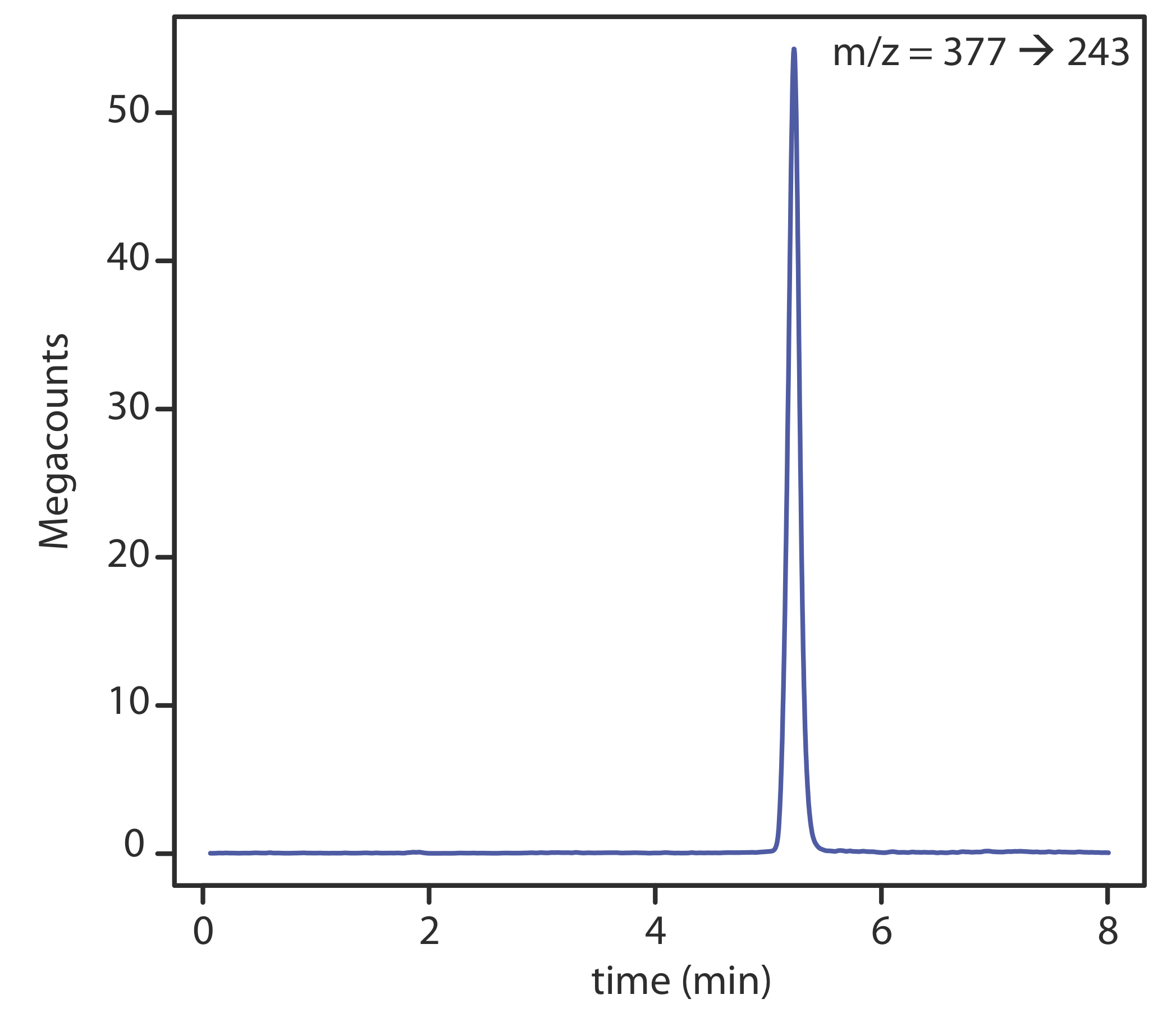
З мас-спектрометром в якості детектора існує кілька варіантів моніторингу хроматограми. Найпоширенішим методом є безперервне сканування всього масового спектру та повідомлення про загальний сигнал для всіх іонів, що досягають детектора під час кожного сканування. Це загальне іонне сканування забезпечує універсальне виявлення для всіх аналітів. Як видно на малюнку Template:index, ми можемо досягти певної міри селективності, контролюючи лише конкретні співвідношення маси до заряду, процес, який називається селективно-іонним моніторингом.
Кількісне застосування перегородкової хроматографії
Роздільна хроматографія зазвичай використовується як для якісного, так і кількісного аналізу екологічних, фармацевтичних, промислових, криміналістичних, клінічних та споживчих зразків продукції.
Підготовка зразків до аналізу
Зразки в рідкій формі вводять у ВЕРХ після відповідного очищення для видалення будь-яких твердих частинок або після відповідної екстракції для видалення матричних перешкод. При визначенні поліароматичних вуглеводнів (ПАГ) у стічних водах, наприклад, екстракція з CH 2 Cl 2 служить подвійному призначенню концентрування аналітів і виділення їх від матричних втручань. Тверді зразки спочатку розчиняють у відповідному розчиннику або аналітах, що представляють інтерес, вносяться в розчин шляхом екстракції. Наприклад, аналіз ВЕРХ для активних інгредієнтів та продуктів розпаду у фармацевтичній таблетці часто починається з вилучення порошкоподібної таблетки з частиною рухомої фази. Зразки газу збирають шляхом барботирования їх через пастку, яка містить відповідний розчинник. Органічні ізоціанати в промислових атмосферах збирають шляхом барботирования повітря через розчин 1- (2-метоксифеніл) піперазину в толуолі. Реакція між ізоціанатами та 1- (2-метоксифеніл) піперазином стабілізує їх проти деградації перед аналізом ВЕРХ і перетворює їх у хімічну форму, яку можна контролювати за допомогою поглинання УФ.
Кількісні розрахунки
Кількісний аналіз ВЕРХ часто простіший, ніж кількісний аналіз ГХ, оскільки петля зразка фіксованого обсягу забезпечує більш точну та точну ін'єкцію. Як результат, більшість кількісних методів ВЕРХ не потребують внутрішнього стандарту і замість цього використовують зовнішні стандарти і нормальну калібрувальну криву.
Внутрішній стандарт необхідний при використанні HPLC-MS, оскільки інтерфейс між ВЕРХ та мас-спектрометром не дозволяє відтворювати передачу елюенту колонки в іонізаційну камеру MS.
Концентрацію поліядерних ароматичних вуглеводнів (ПАГ) у ґрунті визначають шляхом першого вилучення ПАУ метиленхлоридом. Екстракт розбавляється, якщо це необхідно, і ПАУ відокремлюють за допомогою ВЕРХ за допомогою УФ/ВІС або флуоресцентного детектора. Калібрування досягається за допомогою одного або декількох зовнішніх стандартів. У типовому аналізі 2,013-г зразка висушеного ґрунту екстрагують 20,00 мл хлориду метилену. Після фільтрування для видалення грунту видаляють 1,00-мл порцію екстракту і розводять до 10,00 мл ацетонітрилом. Введення 5 мкл розведеного екстракту в ВЕРХ дає сигнал 0,217 (довільні одиниці) для фторантену ПАГ. Коли 5 мкл стандарту фторантену 20,0-ppm аналізується з використанням тих же умов, вимірюється сигнал 0,258. Повідомити частини на мільйон фторантену в грунті.
Рішення
Для одноточкового зовнішнього стандарту взаємозв'язок між сигналом, S, і концентрацією, С, флуорантену становить
\[S = kC \nonumber \]
Підстановка в значеннях сигналу і концентрації стандарту дає значення k як
\[k=\frac{S}{C}=\frac{0.258}{20.0 \text{ ppm}}=0.0129 \text{ ppm}^{-1} \nonumber \]
Використовуючи це значення для k та сигналу ВЕРХ зразка, дає концентрацію фторантену
\[C=\frac{S}{k}=\frac{0.217}{0.0129 \text{ ppm}^{-1}}=16.8 \text{ ppm} \nonumber \]
для витягнутого і розведеного зразка грунту. Концентрація фторантену в грунті становить
\[\frac{16.8 \text{ g} / \mathrm{mL} \times \frac{10.00 \text{ mL}}{1.00 \text{ mL}} \times 20.00 \text{ mL}}{2.013 \text{ g} \text { sample }}=1670 \text{ ppm} \text { fluoranthene } \nonumber \]
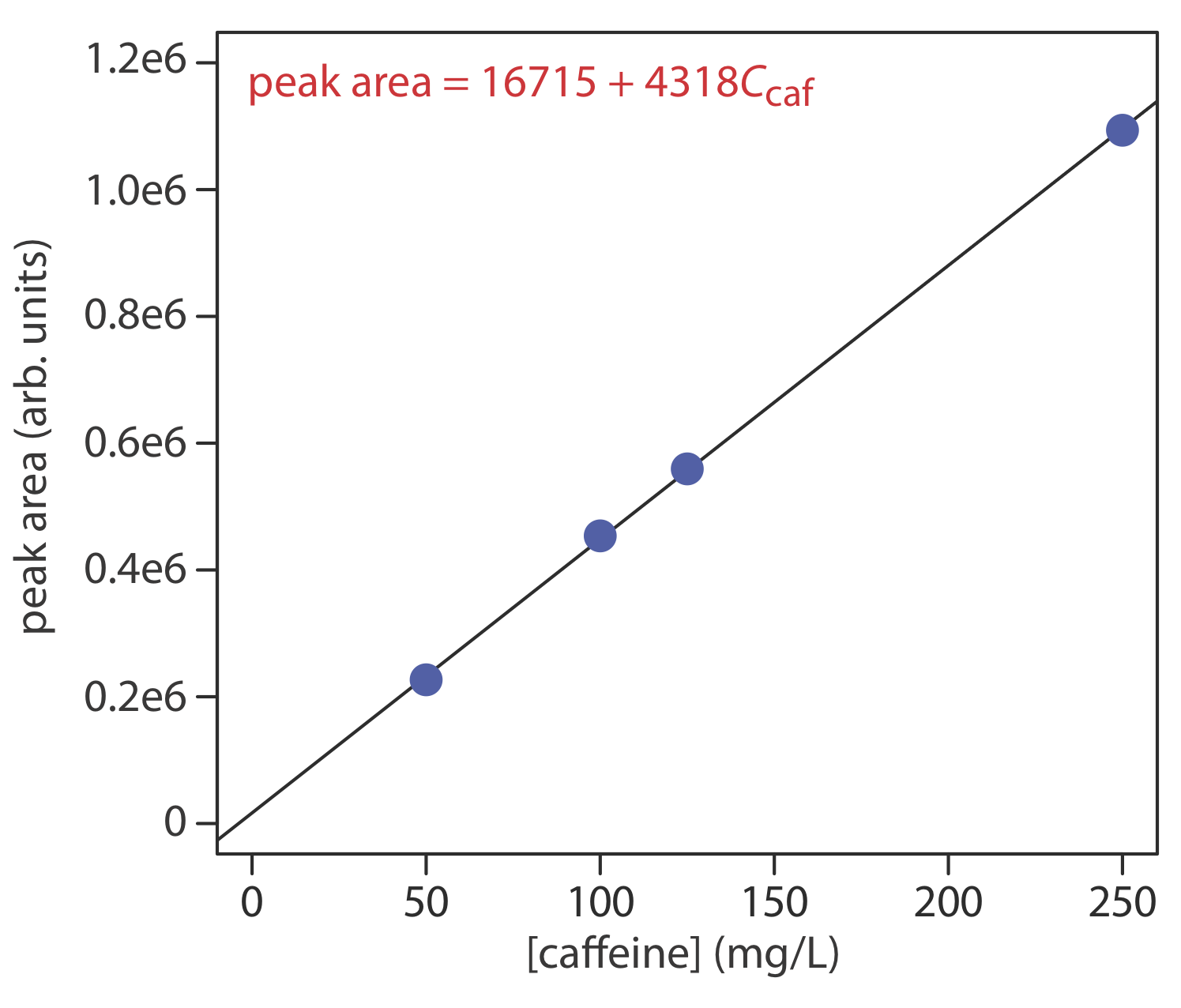
Концентрацію кофеїну в напоях визначають шляхом зворотно-фазового поділу ВЕРХ з використанням рухомої фази 20% ацетонітрилу і 80% води, і за допомогою неполярної колонки С 8. Результати для серії 10-мкл ін'єкцій стандартів кофеїну наведені в наступній таблиці.
| [Кофеїн] (мг/л) | пікова площа (арб. одиниць) |
|---|---|
| 50.0 | 226724 |
| 100.0 | 453762 |
| 125.0 | 559443 |
| 250.0 | 1093637 |
Яка концентрація кофеїну в зразку, якщо ін'єкція 10-мкл дає пікову площу 424195? Дані в цій проблемі походять від Kusch, P.; Knupp, G. «Одночасне визначення кофеїну в напоях Cola та інших напоях за допомогою реверсивної фази HPTLC та ВЕРХ зворотної фази», Chem. Педагог, 2003, 8, 201—205.
- Відповідь
-
На малюнку нижче показана калібрувальна крива і рівняння калібрування для набору зовнішніх стандартів. Заміна пікової площі зразка в калібрувальне рівняння дає концентрацію кофеїну в зразку 94,4 мг/л.