24.3: Кулометрія контрольованого струму
- Page ID
- 27454

Другий підхід до кулометрії полягає у використанні постійного струму замість постійного потенціалу, що призводить до формування профілю струму проти часу, показаного на рис\(\PageIndex{1}\). Кулометрія контрольованого струму має дві переваги перед кулометрією з керованим потенціалом. По-перше, час аналізу коротший, оскільки струм не зменшується з часом. Типовий час аналізу кулометрії з керованим струмом менше 10 хв, у порівнянні з приблизно 30-60 хв для кулометрії з керованим потенціалом. По-друге, оскільки загальний заряд - це просто добуток струму та часу, немає необхідності інтегрувати криву поточного часу на малюнку\(\PageIndex{1}\).
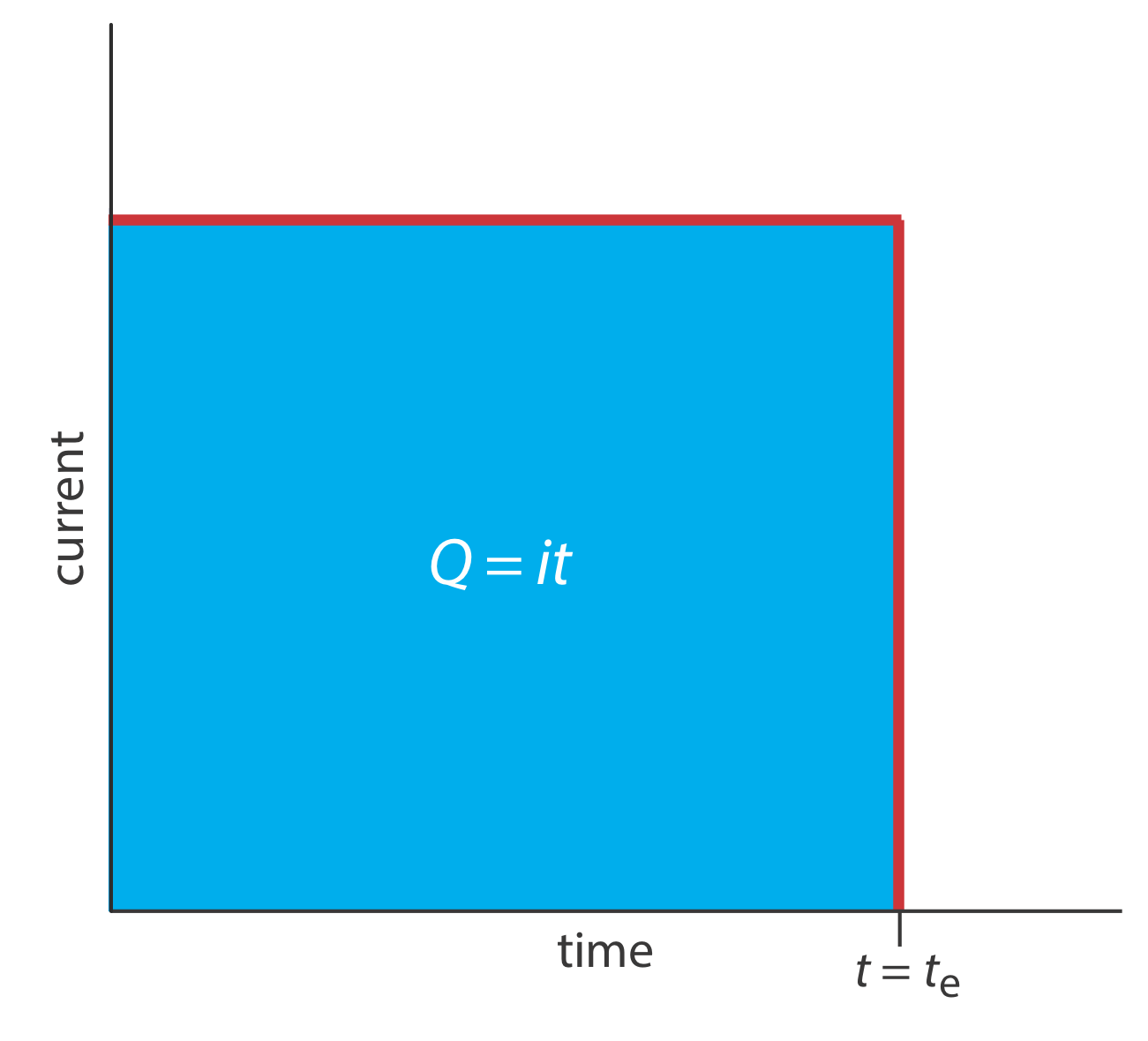
Використання постійного струму представляє нам дві важливі експериментальні проблеми. По-перше, під час електролізу концентрація аналіту - і, отже, струм, що виникає в результаті його окислення або відновлення, постійно зменшується. Для підтримки постійного струму ми повинні дозволити потенціалу змінюватися до тих пір, поки на робочому електроді не відбудеться інша реакція окислення або відновлення. Якщо ми не спроектуємо систему ретельно, ця вторинна реакція призводить до ефективності струму, який менше 100%. Друга проблема полягає в тому, що нам потрібен метод, щоб визначити, коли електроліз аналіта завершений. У кулометричному аналізі з контрольованим потенціалом ми знаємо, що електроліз завершується, коли струм досягає нуля, або коли він досягає постійного фону або залишкового струму. Однак при кулометричному аналізі з контрольованим струмом струм продовжує протікати навіть тоді, коли електроліз аналіта завершений. Необхідний відповідний метод визначення кінцевої точки реакції, т е.
Підтримка поточної ефективності
Щоб проілюструвати, чому зміна потенціалу робочого електрода може привести до ККД струму менше 100%, розглянемо кулометричний аналіз для Fe 2 + на основі його окислення до Fe 3 + при робочому електроді Pt в 1 M H 2 SO 4.
\[\mathrm{Fe}^{2+}(a q) \rightleftharpoons \text{ Fe}^{3+}(a q)+e^{-} \label{ci1} \]
\(\PageIndex{2}\)На малюнку показані відповідні потенціали для цієї системи. На початку аналізу потенціал робочого електрода залишається майже постійним на рівні, близькому до його початкового значення.
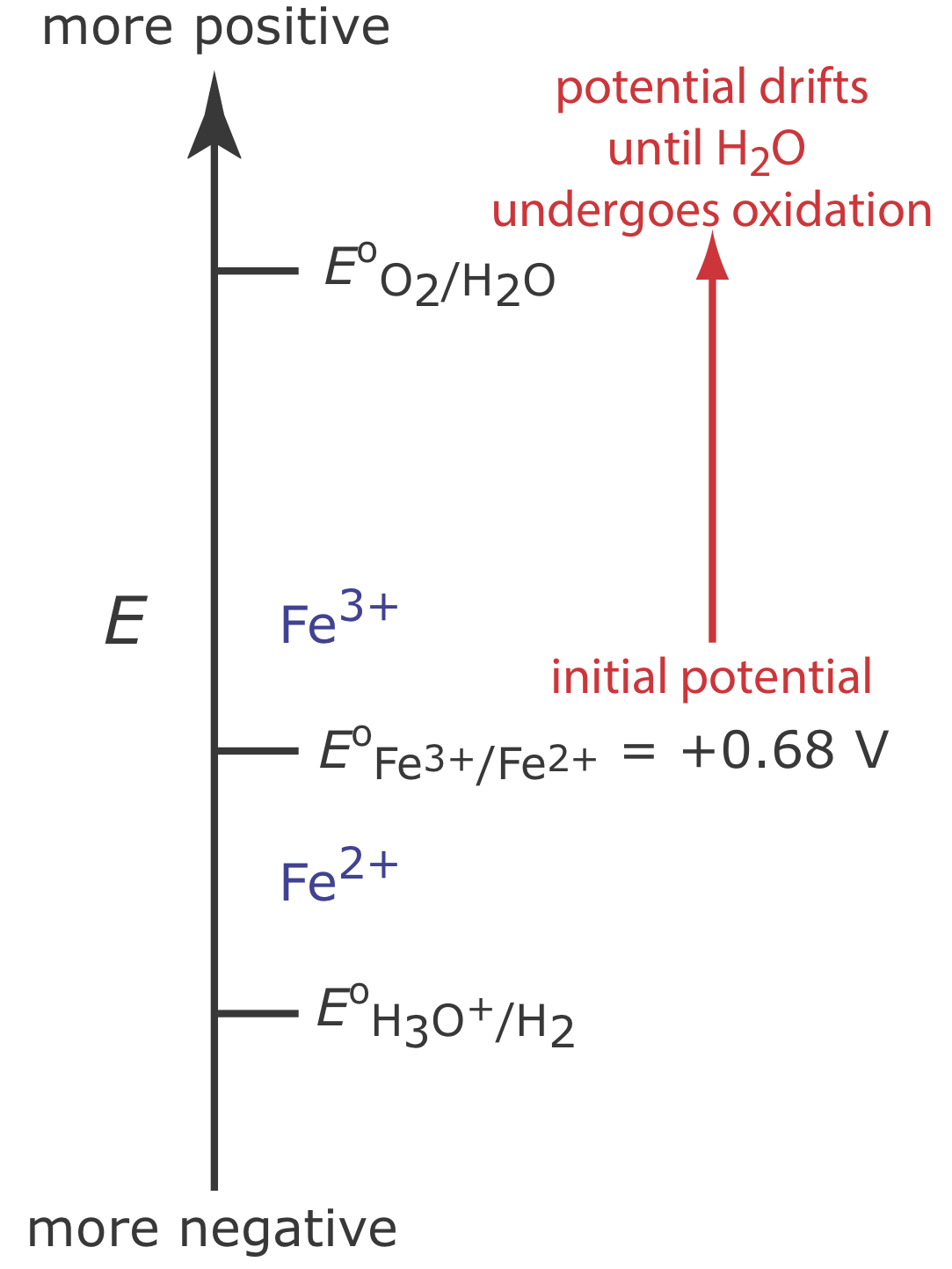
У міру зменшення концентрації Fe 2 + і збільшення концентрації Fe 3 + потенціал робочого електрода зміщується в бік більш позитивних значень, поки не почнеться окислення Н 2 О.
\[2 \mathrm{H}_{2} \mathrm{O}(l)\rightleftharpoons \text{ O}_{2}(g)+4 \mathrm{H}^{+}(a q)+4 e^{-} \label{ci2} \]
Оскільки частина загального струму надходить від окислення H 2 O, ефективність струму для аналізу менше 100%, і ми не можемо використовувати рівняння\(Q = it\) для визначення кількості Fe 2 + у зразку.
Хоча ми не можемо запобігти дрейфу потенціалу, поки інший вид не піддається окисленню, ми можемо підтримувати 100% ефективність струму, якщо продукт цієї реакції вторинного окислення як швидко, так і кількісно реагує з рештою Fe 2 +. Для цього додаємо надлишок Ce 3 + до аналітичного рішення. Як показано на малюнку\(\PageIndex{3}\), при зміщенні потенціалу робочого електрода до більш позитивного потенціалу Ce 3 + починає окислюватися до Се 4 +
\[\mathrm{Ce}^{3+}(a q) \rightleftharpoons \text{ Ce}^{4+}(a q)+e^{-} \label{ci3} \]
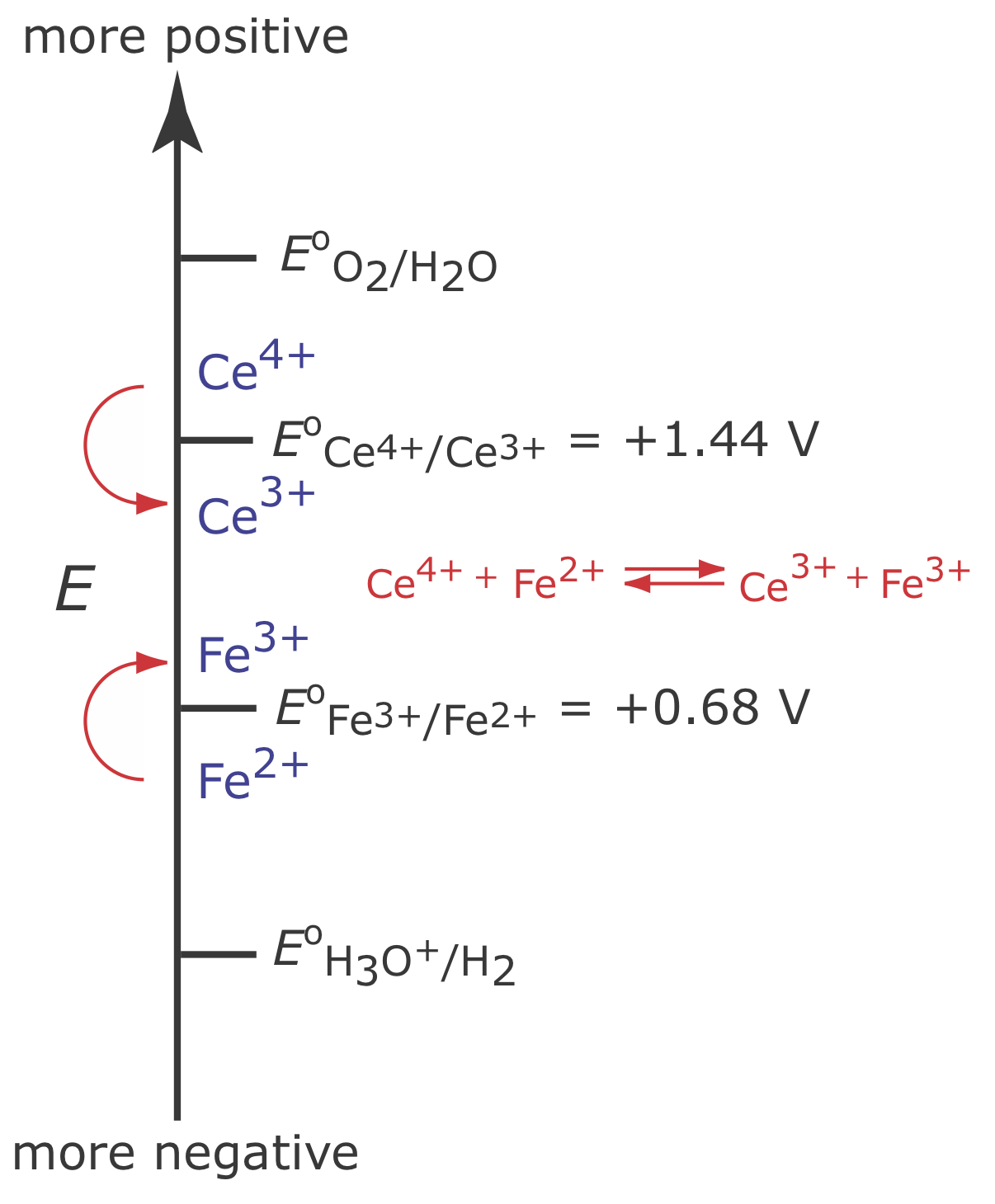
Се 4 +, що утворюється на робочому електроді, швидко змішується з розчином, де вступає в реакцію з будь-яким наявним Fe 2 +.
\[\mathrm{Ce}^{4+}(a q)+\text{ Fe}^{2+}(a q) \rightleftharpoons \text{ Ce}^{3+}(a q)+\text{ Fe}^{3+}(a q) \label{ci4} \]
Поєднання реакції\ ref {ci3} і реакції\ ref {ci4} показує, що чистою реакцією є окислення Fe 2 + до Fe 3 +
\[\mathrm{Fe}^{2+}(a q) \rightleftharpoons \text{ Fe}^{3+}(a q)+e^{-} \label{ci5} \]
який підтримує коефіцієнт корисної дії струму 100%. Вид, який використовується для підтримки 100% ефективності струму, називається медіатором.
Визначення кінцевої точки
Додавання медіатора вирішує проблему збереження 100% ефективності струму, але це не вирішує завдання визначення того, коли електроліз аналіта завершений. Використовуючи аналіз на Fe 2 + на рис.\(\PageIndex{3}\), коли окислення Fe 2 + завершено продовжує надходити струм від окислення Ce 3 +, і, врешті-решт, окислення H 2 O. Що нам потрібно, це сигнал про те, що повідомляє нам, коли в розчині більше немає Fe 2 +.
Для наших цілей зручно розглядати кулометричний аналіз контрольованим струмом як реакцію між аналітом, Fe 2 +, і медіатором Ce 3 +, як показано реакцією\ ref {ci4}. Ця реакція ідентична окислювально-відновному титруванню; таким чином, ми можемо використовувати кінцеві точки для окислювально-відновного титрування - візуальних показників та потенціометричних або кондуктометричних вимірювань - щоб сигналізувати про закінчення кулометричного аналізу контрольованого струму. Наприклад, фероїн забезпечує корисну візуальну кінцеву точку для опосередкованого кулометричного аналізу Ce 3 + для Fe 2 +, змінюючи колір з червоного на синій, коли електроліз Fe 2 + завершений.
Контрольно-вимірювальні прилади
Ми можемо здійснити кулометрію контрольованого струму за допомогою двоелектродного гальваностата, показаного на малюнку\(\PageIndex{4}\), який складається з робочого електрода і зустрічного електрода. Робочий електрод - часто простий електрод Pt - також називають електродом генератора, оскільки саме там медіатор реагує на генерацію виду, який реагує з аналітом. При необхідності зустрічний електрод ізолюють від аналітичного розчину сольовим містком або пористою фриттою, щоб запобігти реакції його продуктів електролізу з аналітом. Струм від джерела живлення через робочий електрод становить
\[i=\frac{E_{\mathrm{PS}}}{R+R_{\mathrm{cell}}} \label{ci6} \]
де Е ПС - потенціал джерела живлення, R - опір резистора, а R осередок - опір електрохімічної осередку. Якщо R >> R осередок, то струм між допоміжним і робочим електродами
\[i=\frac{E_{\mathrm{PS}}}{R} \approx \text{constant} \label{ci7} \]
підтримує постійне значення. Для контролю потенціалу робочого електрода, який змінюється в міру зміни складу електрохімічної комірки, ми можемо включити додатковий опорний електрод і потенціометр з високим імпедансом.

Крім того, ми можемо генерувати окислювач або відновник зовні і дозволити йому текти в аналітичний розчин. \(\PageIndex{5}\)На малюнку показаний один простий метод для цього. Розчин, що містить медіатор, перетікає в електрохімічну комірку невеликого об'єму, продукти виходять через окремі трубки. Залежно від аналіту окислювач або відновлювальний реагент доставляється в аналітичний розчин. Наприклад, ми можемо генерувати Ce 4 + за допомогою водного розчину Ce 3 +, направляючи Ce 4 +, який утворюється на аноді, до нашого зразка.
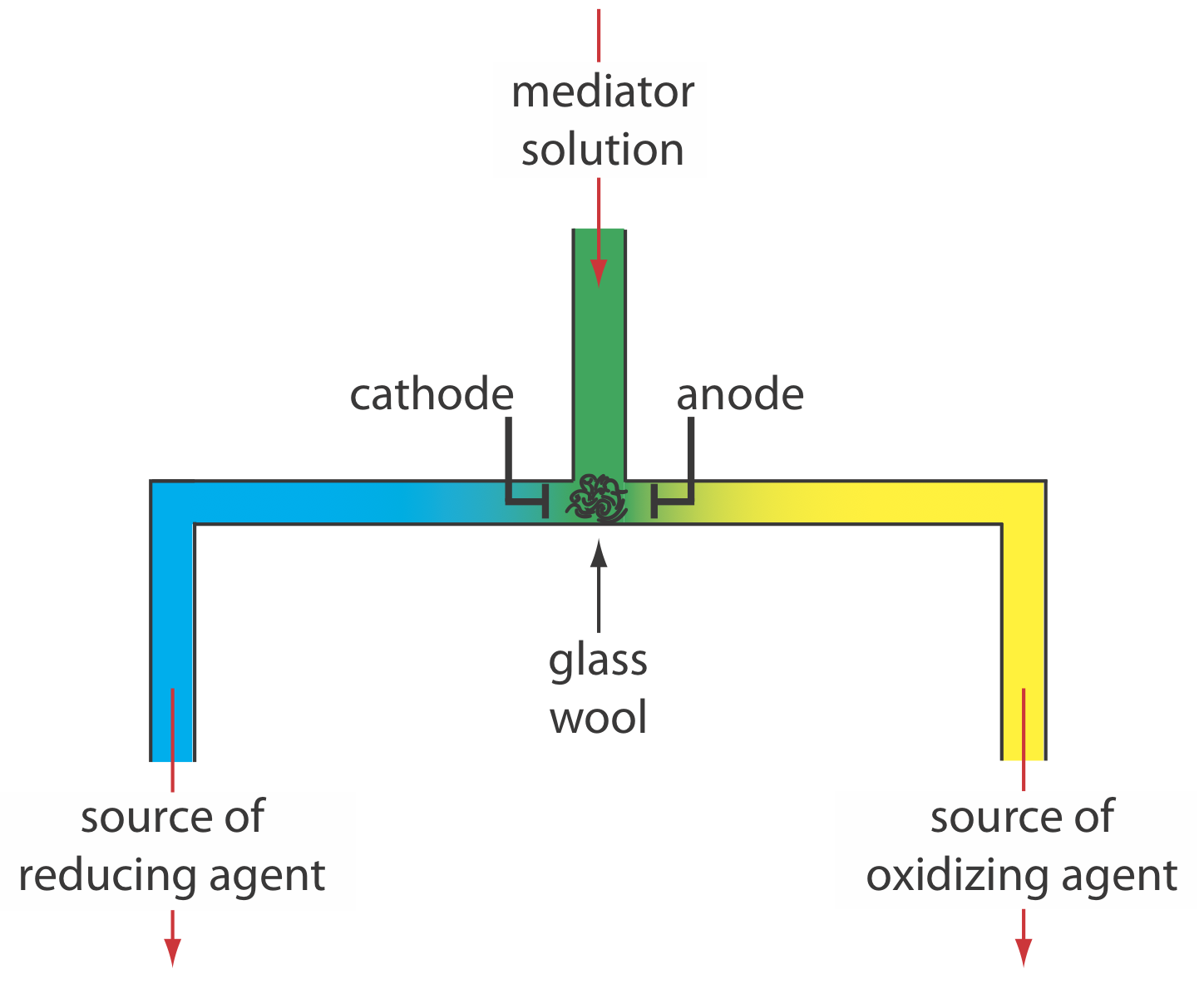
Є ще дві найважливіші потреби в кулометрії контрольованого струму: точний годинник для вимірювання часу електролізу, т е, і перемикач для запуску і зупинки електролізу. Аналоговий годинник може записувати час до найближчого ± 0,01 с, але необхідність зупинки та запуску електролізу при наближенні до кінцевої точки може призвести до загальної невизначеності ± 0,1 с Цифровий годинник дозволяє більш точно виміряти час із загальною невизначеністю ± 1 мс. Перемикач повинен контролювати як струм, так і годинник, щоб ми могли зробити точне визначення часу електролізу.
Кулометричні титрування
Кулометричний метод контрольованого струму іноді називають кулометричним титруванням через його схожість зі звичайним титруванням. Наприклад, в кулометричному аналізі контрольованого струму для Fe 2 + за допомогою медіатора Ce 3 + окислення Fe 2 + Ce 4 + (реакція\ ref {ci4}) ідентично реакції при окислювально-відновному титруванні.
Є й інші подібності між кулометрією з контрольованим струмом і титриметрією. Якщо об'єднати рівняння\(Q = nFN_a\) і рівняння\(Q = it_e\) і вирішити для молей аналіта N A, то отримаємо наступне рівняння.
\[N_{A}=\frac{i}{n F} \times t_{e} \label{ci8} \]
Порівняйте рівняння\ ref {ci8} із співвідношенням між молями аналіту, N A та молями титранту, N T, у титруванні
\[N_{A}=N_{T}=M_{T} \times V_{T} \label{ci9} \]
де M T і V T - молярність титранта і об'єм титранту в кінцевій точці. У кулометрії з постійним струмом джерело струму еквівалентно титранту, і значення цього струму аналогічно молярності титранта. Час електролізу аналогічний об'єму титранту, а t e еквівалентно кінцевій точці титрування. Нарешті, перемикач пуску і зупинки електролізу виконує ту ж функцію, що і запірний кран бюрета.
Для простоти ми припустили вище, що стехіометрія між аналітом і титрантом дорівнює 1:1. Однак припущення не є важливим і не впливає на наше спостереження за подібністю між кулометрією з контрольованим струмом і титруванням.
Кількісні програми
Використання медіатора робить кулометричне титрування більш універсальним аналітичним прийомом, ніж кулометрія з керованим потенціалом. Наприклад, пряме окислення або відновлення білка на робочому електроді важко, якщо активний окислювально-відновний ділянку білка лежить глибоко в його структурі. Кулометричне титрування білка можливо, однак, якщо ми використовуємо окислення або відновлення медіатора для отримання виду розчину, який реагує з білком. Таблиця\(\PageIndex{1}\) узагальнює кілька кулометричних методів контрольованого струму, заснованих на окислювально-відновній реакції з використанням медіатора.
Для аналіту, який нелегко окислювати або зменшити, ми можемо завершити кулометричне титрування шляхом з'єднання окислення або відновлення медіатора з кислотно-основою, осадженням або реакцією комплексоутворення, яка включає аналіт. Наприклад, якщо ми використовуємо H 2 O як медіатор, ми можемо генерувати H 3 O + на аноді
\[6 \mathrm{H}_{2} \mathrm{O}(l) \rightleftharpoons 4 \mathrm{H}_{3} \text{O}^{+}(a q)+\text{ O}_{2}(g)+4 e^{-} \nonumber \]
і генерувати OH — на катоді.
\[2 \mathrm{H}_{2} \mathrm{O}(l)+2 e^{-} \rightleftharpoons 2 \mathrm{OH}^{-}(a q)+\text{ H}_{2}(g) \nonumber \]
Якщо ми проводимо окислення або відновлення Н 2 О за допомогою генераторної осередку на рис.\(\PageIndex{5}\), то ми можемо вибірково дозувати Н 3 О + або ОН — в розчин, який містить аналіт. Отримана реакція ідентична реакції при кислотно-лужному титруванні. Кулометричні кислотно-лужні титрування використовувались для аналізу сильних і слабких кислот і основ як у водних, так і в неводних матрицях. Таблиця\(\PageIndex{2}\) узагальнює кілька прикладів кулометричних титрувань, які включають реакції кислотно-основи, комплексоутворення та осадження.
У порівнянні зі звичайним титруванням, кулометричне титрування має дві важливі переваги. Перша перевага полягає в тому, що електрохімічна генерація титранту дозволяє нам використовувати нестійкий реагент. Хоча ми не можемо приготувати та зберігати розчин високореактивного реагенту, такого як Ag 2 + або Mn 3 +, ми можемо генерувати їх електрохімічним шляхом та використовувати їх в кулометричному титруванні. По-друге, оскільки відносно легко виміряти невелику кількість заряду, ми можемо використовувати кулометричне титрування для визначення аналіту, концентрація якого занадто мала для звичайного титрування.
Наступний приклад показує розрахунки для типового кулометричного аналізу.
Для визначення чистоти зразка Na 2 S 2 O 3 зразок титрують кулометрично, використовуючи I — як медіатор і\(\text{I}_3^-\) як титрант. Пробу вагою 0,1342 г переносять в об'ємну колбу об'ємом 100 мл і розводять до об'єму дистильованою водою. На електрохімічну комірку переносять порцію 10,00 мл разом з 25 мл 1 М КІ, 75 мл фосфатного буфера pH 7,0 і декількома краплями розчину індикатора крохмалю. Електроліз при постійному струмі 36,45 мА вимагає 221,8 с для досягнення кінцевої точки показника крохмалю. Визначте чистоту зразка.
Рішення
Як показано в таблиці\(\PageIndex{1}\), кулометричне титрування\(\text{S}_2 \text{O}_3^{2-}\) з\(\text{I}_3^-\) дорівнює
\[2 \mathrm{S}_{2} \mathrm{O}_{3}^{2-}(a q)+\text{ I}_{3}^{-}(a q)\rightleftharpoons \text{ S}_{4} \mathrm{O}_{6}^{2-}(a q)+3 \mathrm{I}^{-}(a q) \nonumber \]
Для окислення\(\text{S}_2 \text{O}_3^{2-}\) до\(\text{S}_4 \text{O}_6^{2-}\) потрібно один електрон на\(\text{S}_2 \text{O}_3^{2-}\) (n = 1). Поєднання рівнянь\(Q = nFN_A\) і\(Q = it_e\), і рішення для молів і грам Na 2 S 2 O 3 дає
\[N_{A} =\frac{i t_{e}}{n F}=\frac{(0.03645 \text{ A})(221.8 \text{ s})}{\left(\frac{1 \text{ mol } e^{-}}{\text{mol Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3}}\right)\left(\frac{96487 \text{ C}}{\text{mol } e^{-}}\right)} =8.379 \times 10^{-5} \text{ mol Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3} \nonumber \]
Це кількість Na 2 S 2 O 3 в 10,00-мл порції 100-мл зразка; таким чином, в вихідному зразку є 0,1325 грам Na 2 S 2 O 3. Таким чином, чистота зразка
\[\frac{0.1325 \text{ g} \text{ Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3}}{0.1342 \text{ g} \text { sample }} \times 100=98.73 \% \text{ w} / \text{w } \mathrm{Na}_{2} \mathrm{S}_{2} \mathrm{O}_{3} \nonumber \]
Відзначимо, що для такого розрахунку не має значення,\(\text{S}_2 \text{O}_3^{2-}\) окислюється чи на робочому електроді або окислюється шляхом\(\text{I}_3^-\).