7: Вирівнювання послідовності
- Page ID
- 66620

Програма BLAST (Basic Local Alignment Search Tool) використовує алгоритми вирівнювання послідовності для порівняння послідовності запитів з базою даних для ідентифікації інших відомих послідовностей, подібних до послідовності запитів. Часто анотації, прикріплені до вже відомих послідовностей, дають важливу біологічну інформацію про послідовність запитів. Практично всі біологи використовують BLAST, роблячи вирівнювання послідовностей одним з найважливіших алгоритмів біоінформатики.
Досліджувана послідовність може складатися з нуклеотидів (з ДНК нуклеїнових кислот або РНК) або амінокислот (з білків). Нуклеїнові кислоти з'єднують між собою чотири різних нуклеотиди: A, C, T, G для ДНК і A, C, U, G для РНК; білки з'єднують між собою двадцять різних амінокислот. Послідовність молекули ДНК або білка - це лінійний порядок нуклеотидів або амінокислот у заданому напрямку, який визначається хімією молекули. Нам не потрібно знати точні деталі хімії; достатньо знати, що білок має помітні кінці, звані N-кінцевим і C-кінцем, і що звичайною умовою є читання послідовності амінокислот від N-кінця до C-кінця. Специфікація напрямку є більш складною для молекули ДНК, ніж для білкової молекули через подвійну спіраль структури ДНК, і це буде пояснено в розділі 7.1.
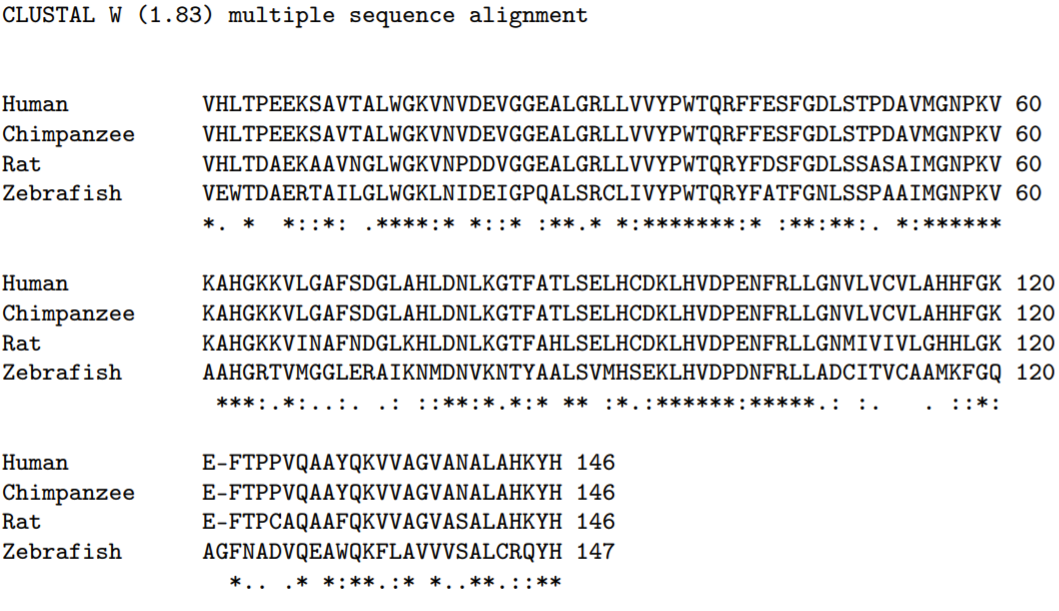
Основний алгоритм вирівнювання послідовностей вирівнює дві або більше послідовностей, щоб виділити їх схожість, вставляючи невелику кількість прогалин у кожну послідовність (зазвичай позначається тире), щоб вирівняти, де це можливо, однакові або подібні символи. Наприклад, Рис\(7.1\) представляє вирівнювання за допомогою програмного інструменту ClusTalW гемоглобіну бета-ланцюга від людини, шимпанзе, щура та зебрариби. Послідовності людини та шимпанзе ідентичні, наслідок наших дуже тісних еволюційних відносин. Послідовність щурів відрізняється від людини/шимпанзе лише 27 з 146 амінокислот; всі ми ссавці. Послідовність зебрафіш, хоч і чітко пов'язана, значно розходиться. Зверніть увагу на введення розриву в кожній з послідовностей ссавців в позиції амінокислоти зебри риби 122. Це дозволяє подальшій послідовності зебрафіш краще узгоджуватися з послідовностями ссавців і передбачає або введення нової амінокислоти в рибу, або видалення амінокислоти у ссавців. Вставка або видалення символу в послідовності називається інделем. Розбіжності в послідовності, такі як те, що відбувається між зебрарибами і ссавцями в амінокислотних позиціях 2 і 3, називається мутацією. ClastAlw розміщує «*» на останньому рядку, щоб позначити точні збіги амінокислот у всіх послідовностях,\(\mathrm{a}^{\prime}: '\) і «\('\)'для позначення хімічно подібних амінокислот у всіх послідовностях (кожна амінокислота має характерні хімічні властивості, і амінокислоти можуть бути згруповані відповідно до аналогічних властивостей). У цьому розділі ми детально описуємо алгоритми, які використовуються для вирівнювання послідовностей.
- 7.2: Вирівнювання грубої сили
- Один (поганий) підхід до вирівнювання послідовностей полягає у вирівнюванні двох послідовностей усіма можливими способами, оцінка вирівнювання за передбачуваною системою балів та визначення найвищого вирівнювання балів. Проблема цього підходу грубої сили полягає в тому, що кількість можливих вирівнювань зростає експоненціально з довжиною послідовності; а для послідовностей розумної довжини обчислення вже неможливо.
- 7.3: Динамічне програмування
- Дві послідовності розумних розмірів не можуть бути вирівняні грубою силою. На щастя, існує ще один алгоритм, запозичений з інформатики, динамічне програмування, який використовує динамічну матрицю.
- 7.4: Прогалини
- Емпіричні дані свідчать про те, що розриви кластерні, як в нуклеотидних, так і в білкових послідовностях. Кластеризація зазвичай моделюється різними штрафами за відкриття зазору.
- 7.5: Локальні вирівнювання
- Ми досі обговорювали, як вирівняти дві послідовності по всій їх довжині, що називається глобальним вирівнюванням. Однак часто корисніше вирівняти дві послідовності лише над частиною їх довжин, званих локальним вирівнюванням. У біоінформатиці алгоритм глобального вирівнювання називається «Needleman-Wunsch», а що для локального вирівнювання «Сміт-Уотерман».
- 7.6: Програмне забезпечення
- Якщо у вас є в руках дві або більше послідовностей, які ви хотіли б вирівняти, є вибір програмних інструментів доступні.