4.1: Кінетика і механізми-
- Page ID
- 25207

Одним з найпотужніших джерел доказів того, як протікає та чи інша реакція, є вивчення швидкості реакції або кінетики реакції. У таких дослідженнях, як змінюється швидкість реакції [1], вимірюється в залежності від концентрації кожного реагенту. Одним з найпоширеніших способів вимірювання цієї зміни є використання спектроскопічної техніки. Наприклад, якщо з'єднання поглинає в області UV-VIS спектра, поглинання пропорційно концентрації. Тому, якщо концентрація речовини змінюється, її можна виміряти змінами поглинання. Реакцію проводять кілька разів з усіма, крім одного з реагентів, встановленими постійними; потім додається інша концентрація решти реагенту та вимірюється швидкість реакції. Це повторюється для кожного реагенту протягом декількох концентрацій. Кінцевим результатом такого дослідження є отримання того, що відоме як рівняння швидкості; для загальної реакції\(\mathrm{A}+\mathrm{B} \rightarrow \mathrm{C}+\mathrm{D}\) рівняння швидкості набуває вигляду:\[\text { Rate }=k[\mathrm{A}]^{x}[\mathrm{B}]^{y}\]
У цьому рівнянні\(k\) є постійною швидкості, а показники x і y розповідають нам про те, як концентрація кожного реагенту впливає на швидкість. Сума показників (тобто\(x+y+\ldots\)) - це порядок реакції. Наприклад, якщо\(x = 1\), то норма безпосередньо пов'язана з концентрацією\(\mathrm{A}\). Якщо обидва\(x\) і\(y = 1\), то швидкість прямо пропорційна обом\([\mathrm{A}]\) і\([\mathrm{B}]\), а загальний порядок реакції дорівнює = 2. Якщо показник = 0, то швидкість не залежить від цієї концентрації реагенту, і цей реагент можна видалити з рівняння закону швидкості (оскільки\([n]^{0}=1\) незалежно від того, яке значення концентрації n).
Найважливіша ідея, яку слід пам'ятати, полягає в тому, що рівняння швидкості містить лише реагенти, які беруть участь у кроці визначення швидкості (тобто найповільнішому кроці) реакції. Якщо реакція протікає по ряду ступенів, то крок з найбільшою енергією активації буде нормовизначальним, і в нормовому законі будуть присутні тільки ті реагенти, які беруть участь в цьому кроці. [2] Оскільки закон ставки визначається емпіричним шляхом, закон ставки надає нам докази про механізм реакції.
Докази для\(\mathbf{S}_{\mathbf{N}} 2\) механізму:
Реакція, яку ми обговорювали раніше в курсі, відома як\(\mathrm{S}_{\mathrm{N}} 2\) реакція, яка є скороченою для S заміни, N нуклеофілічна, другого порядку. Ми запропонували механізм цієї реакції, не надаючи жодних емпіричних доказів, але тепер давайте скористаємося деякими з того, що ви навчилися, щоб більш ретельно розглянути докази цього механізму.
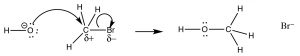
Реакція другого порядку:
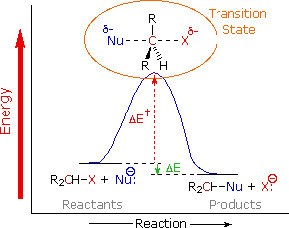
перший доказ походить від закону кінетичної швидкості. Швидкість реакції залежить як від концентрації субстрату, так і від нуклеофіла:\(\text { rate }=k[\mathrm{RX}][\mathrm{Nu}]\). Це означає, що обидва повинні бути присутніми на кроці визначення ставки. Найпростіше пояснення, яке узгоджується з цим висновком, - це те, що ми вже запропонували: нуклеофіл атакує електрофільний вуглець одночасно з виходом групи. Тобто реакція відбувається в один безперервний етап. Діаграма енергії реакції, на якій ми будуємо прогрес реакції Energy v, виглядає так (\(\rightarrow\)). У цій реакції є тільки один енергетичний бар'єр, тільки один максимум в шляху реакції. Енергія цього бар'єру відома як енергія активації\(\Delta \mathrm{E}_{+}^{+}\). Дивлячись на діаграму реакції, також відзначимо, що реакція екзотермічна (або ексергонічна, якщо ми будуємо енергію Гіббса), так як загальна реакція негативна).\(\Delta \mathrm{E}\) Вид на піку активації енергетичного бар'єру відомий як перехідний стан, а його структура і пов'язана енергія визначає швидкість реакції.
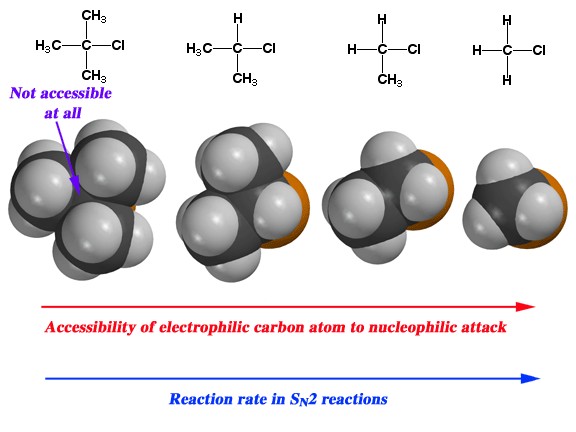
На структуру субстрату впливає норма:
Можливо, ви помітили в наших попередніх обговореннях нуклеофільної заміщення, що органічний субстрат завжди був або метилом, або первинним вуглецем, прикріпленим до хорошої групи, що йде. Причина полягала в тому, що в розглянутих нами реакціях і швидкість, і механізм реакції сильно залежать від структури субстрату. Зі збільшенням кількості метильних груп, прикріплених до первинного вуглецю (від 0 для самої метильної групи до трьох [третинних]) швидкість реакції сповільнюється, як показано. Швидкість\(\mathrm{S}_{\mathrm{N}} 2\) реакції на третинний субстрат незначна.
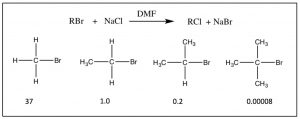
Отже, виникають два питання: по-перше, чому ці швидкості реакції відрізняються? і по-друге, чому ця зміна швидкості реакції свідчить про\(\mathrm{S}_{\mathrm{N}} 2\) механізм? На обидва можна відповісти, придивившись до реакції з молекулярної точки зору. Пам'ятайте, що всі реагенти розчиняються в розчиннику; тепловий рух призводить до їх зіткнення один з одним і з молекулами розчинника. Щоб нуклеофіл і субстрат реагували один з одним, спочатку їм доводиться стикатися один з одним. Щоб реакція відбулася, це зіткнення повинно передавати достатньо енергії, щоб комплекс (субстрат + нуклеофіл) міг сформувати перехідний стан - крім того, щоб сформувати молекулу перехідного стану, молекули повинні стикатися один з одним у правильній орієнтації. Після утворення перехідний стан може розпастися з утворенням продуктів реакції.
Нагадаємо, що запропонована нами структура для перехідного стану для цієї реакції має центральний вуглець, з'єднаний з п'ятьма групами: вхідним нуклеофілом, що йде групою і трьома іншими замісниками, які не змінюються під час реакції (вони не є частиною реакції). Оскільки зв'язок утворюється між нуклеофілом і субстратом вуглецю, а зв'язок розривається між вуглецем і вихідною групою, вуглець змінює свій стан гібридизації. Що це означає? У молекулі субстрату реагуючий вуглець прикріплюється до оточуючих груп (\(\mathrm{H-}\)або\(\left.\mathrm{CH}_{3}-\right)\)) зв'язками, утвореними з\(\mathrm{sp}^{3}\) орбіталей. У перехідному стані цей вуглець ще приєднується до тих груп, які залишаться в молекулі продукту, але тепер з зв'язками, утвореними з\(\mathrm{sp}^{2}\) орбіталей. Крім того, він все ще прикріплений як до виїжджаючої групи, так і до вхідного нуклеофіла, використовуючи орбітальну p для формування цих часткових зв'язків. Ви можете думати про цей процес як електронну щільність, що направляється від нуклеофіла через вуглець і з іншого боку до групи, що йде. Однак для цього нуклеофіл може почати зв'язуватися лише тоді, коли він наближається із задньої частини зв'язку до групи, що виходить\(\rightarrow\).
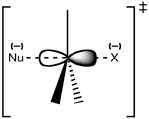
У цей момент ви цілком можете запитати: яке все це має відношення до структури субстрату? Для реакції єдиними продуктивними зіткненнями є ті, де нуклеофіл починає утворювати зв'язок із задньою частиною\(\mathrm{sp}^{3}\) гібридної орбіти; але структура субстрату впливає на ймовірність такої події. У третинних субстратах (наприклад\(\left(\mathrm{CH}_{3}\right)_{3} \mathrm{CBr}\)) наближенню до субстрату перешкоджають громіздкі алкільні групи, такі, що ймовірність взаємодії нуклеофіла з реактивним центром низька. Це явище називається стеричним перешкодою і дає пояснення порядку реакції\(\mathrm{S}_{\mathrm{N}} 2\) реакцій.
\(\mathrm{S}_{\mathrm{N}} 2\)реакції в хіральному центрі:
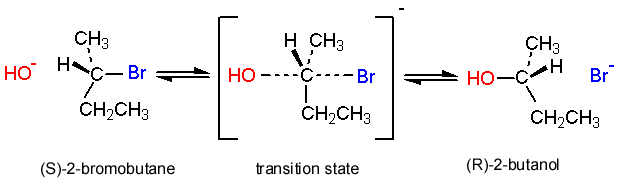
Ще одним доказом\(\mathrm{S}_{\mathrm{N}} 2\) механізму є те, що відбувається, коли\(\mathrm{S}_{\mathrm{N}} 2\) реакція відбувається в хіральному центрі (всередині молекули). Виявляється, конфігурація в цьому центрі змінюється; вуглець інвертується (як парасолька, що дме навиворіт на вітрі), так що S енантіомер перетворюється на енантіомер R. Насправді можна стежити за ходом реакції S N 2 за участю хірального центру за допомогою поляриметра (приладу, який використовується для вимірювання оптичної активності); коли реакція переходить до завершення, оптичне обертання розчину змінюється з часом. Для кожної конкретної підкладки напрямок і величина обертання для виробу будуть різними. Це явище називається інверсією Уолдена і надає ще один доказ для підтримки запропонованого механізму реакції.
Роль розчинника в\(\mathrm{S}_{\mathrm{N}} 2\) реакції:
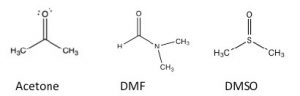
\(\mathrm{S}_{\mathrm{N}} 2\)реакції зазвичай проводяться в розчиннику (чому це?). Емпіричні дослідження показують, що такі реакції протікають швидше, коли проводяться в так званому полярному апротонному розчиннику. Так що ж таке полярний апротний розчинник? Термін означає, що розчинник полярний, але без кислих протонів. Прикладами полярних апротних розчинників є ацетон, диметилформамід (ДМФ) та диметилсульфоксид (ДМСО): кожен є полярним, але не вистачає потенційно кислого протона, такого як Н, який пов'язаний з етанолом\(\mathrm{CH}_{3} \mathrm{CH}_{2} \mathrm{OH}\) або у воді\(\mathrm{H-O-H}\).\(\mathrm{O}\) Вода (і метанол і етанол) є полярним протонним розчинником. У полярному апротонному розчиннику негативний кінець\(\mathrm{C=O}\) або\(\mathrm{S=O}\) диполь локалізується на\(\mathrm{O}\), тоді як позитивний кінець дифузний і делокалізований. Наприклад, в ацетоні кисень має\(\delta-\) заряд на кисень, тоді як позитивний заряд диполя делокалізується як над метильними групами, так\(\mathrm{C}\) і над метильними групами, як показано на карті електростатичного потенціалу (\(\rightarrow\)). На практиці полярні апротичні розчинники можуть добре сольватувати катіони через взаємодію з локалізованим негативним кінцем диполя, але вони не можуть дуже добре сольватувати аніони.
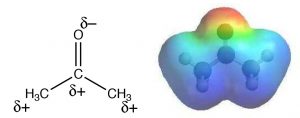
Нагадаємо, що сольватія - це взаємодія, що знижує енергію системи, роблячи її більш стабільною (менш реактивної). Тому розчинник, який залишає нуклеофіл (аніон) нерозчиненим, зробить його більш реактивним. На відміну від цього, полярний протичний розчинник (наприклад, вода або етанол) може сольватувати нуклеофіл через взаємодію з позитивним кінцем диполя, який локалізується на кислому\(\mathrm{H}\), стабілізуючи нуклеофіл і роблячи його менш реактивним. \(\mathrm{S}_{\mathrm{N}} 2\)Підсумовуючи, реакції відбуваються в один етап з інверсією в хіральному центрі. Такі реакції, як правило, швидші для безперешкодних субстратів і прискорюються при проведенні в полярних апротичних розчинниках.