10.4: Атомно-абсорбційна спектроскопія
- Page ID
- 24972

Гайстав Кірхофф і Роберт Бунсен вперше використовували атомне поглинання - разом з атомними викидами - у 1859 та 1860 роках як засіб для ідентифікації атомів у полум'ї та гарячих газах. Хоча атомна емісія продовжувала розвиватися як аналітична техніка, прогрес в атомному поглинанні нудився майже століття. Сучасна атомно-абсорбційна спектроскопія бере свій початок в 1955 році в результаті самостійної роботи А.К. Уолш і К.Т. J. Alkemade [(a) Уолш, А.анал. Хім. 1991, 63, 933А—941А; (б) Коіртіоганн, С.Р. анал. Хім. 1991, 63, 1024—1031А; (с) Славін, В. анал. Хім. 1991, 63, 1033—1038А]. Комерційні інструменти були встановлені на початку 1960-х років, і важливість атомного поглинання як аналітичного методу незабаром була очевидною.
Контрольно-вимірювальні прилади
Атомно-абсорбційні спектрофотометри використовують ту ж однопроменеву або двопроменеву оптику, описану раніше для спектрофотометрів молекулярного поглинання (див. Рис. 10.3.2 і рис. 10.3.3). Однак існує важлива додаткова потреба в атомно-абсорбційній спектроскопії: спочатку ми повинні приховати аналіт у вільні атоми. У більшості випадків аналіт знаходиться у формі розчину. Якщо проба тверда речовина, то перед аналізом ми повинні внести аналіт в розчин. При аналізі осадового осаду на Cu, Zn і Fe, наприклад, ми вводимо аналіти в розчин як Cu 2 +, Zn 2 +, і Fe 3 + шляхом екстрагування їх відповідним реагентом. З цієї причини в даному розділі розглядається тільки введення зразків розчину.
Який реагент ми вирішили використовувати для введення аналіту в розчин, залежить від наших цілей дослідження. Якщо нам потрібно знати загальну кількість металу в осаду, то ми можемо спробувати мікрохвильове травлення, використовуючи суміш концентрованих кислот, таких як HNO 3, HCl та HF. Це руйнує матрицю осаду і приводить все в розчин. З іншого боку, якщо наш інтерес є біологічно доступними металами, ми можемо витягти зразок в більш м'яких умовах, використовуючи, наприклад, розведений розчин HCl або CH 3 COOH при кімнатній температурі.
Розпилення
Процес перетворення аналіту у вільний газоподібний атом називається атомізацією. Перетворення водного аналіту у вільний атом вимагає, щоб ми видалили розчинник, випаровували аналіт і, якщо необхідно, дисоціювали аналіт на вільні атоми. Наприклад, зневоднення водного розчину CuCl 2 залишає нас твердими частинками CuCl 2. Перетворення твердих частинок CuCl 2 в атоми газових фаз Cu і Cl вимагає теплової енергії.
\[\mathrm{CuCl}_{2}(a q) \rightarrow \mathrm{CuCl}_{2}(s) \rightarrow \mathrm{Cu}(g)+2 \mathrm{Cl}(g) \nonumber\]
Існує два загальні методи розпилення: розпилення полум'я та електротермічне розпилення, хоча деякі елементи розпилюються за допомогою інших методів.
Полум'я Атомайзер
На малюнку Template:index показана типова збірка розпилення полум'я з крупним планом декількох ключових компонентів. У показаному тут агрегаті водний зразок втягується в вузол, пропускаючи струмінь стисненого повітря високого тиску повз кінець капілярної трубки, зануреної в зразок. Коли зразок виходить з небулайзера, він б'є скляну ударну кульку, яка перетворює його в дрібний аерозольний туман всередині розпилювальної камери. Аерозольний туман змітається через камеру розпилення газами згоряння - стисненим повітрям та ацетиленом в цьому випадку - до головки пальника, де теплова енергія полум'я десольтує аерозольний туман до сухого аерозолю дрібних твердих частинок. Потім теплова енергія полум'я випаровує частинки, утворюючи пар, який складається з молекулярних видів, іонних видів та вільних атомів.

пальник. Пальник слота на рисунку Template:index a забезпечує довгу оптичну довжину шляху і стабільне полум'я. Оскільки поглинання прямо пропорційне довжині шляху, довга довжина шляху забезпечує більшу чутливість. Стабільне полум'я мінімізує невизначеність через коливання полум'я.
Пальник монтується на регульованої щаблі, що дозволяє всьому вузлу рухатися по горизонталі і вертикалі. Горизонтальні регулювання забезпечують вирівнювання полум'я з оптичним шляхом приладу. Вертикальні регулювання змінюють висоту всередині полум'я, з якого контролюється поглинання. Це важливо, оскільки два конкуруючих процесу впливають на концентрацію вільних атомів у полум'ї. Чим більше часу аналіт проводить у полум'ї, тим більша ефективність розпилення; таким чином, виробництво вільних атомів збільшується з висотою. З іншого боку, більш тривалий час перебування дозволяє більше можливостей для вільних атомів поєднуватися з киснем, утворюючи молекулярний оксид. Як видно на малюнку Template:index, для металу це легко окислюється, наприклад Cr, концентрація вільних атомів найбільша трохи вище головки пальника. Для металу, такого як Ag, який важко окислюється, концентрація вільних атомів неухильно зростає з висотою.
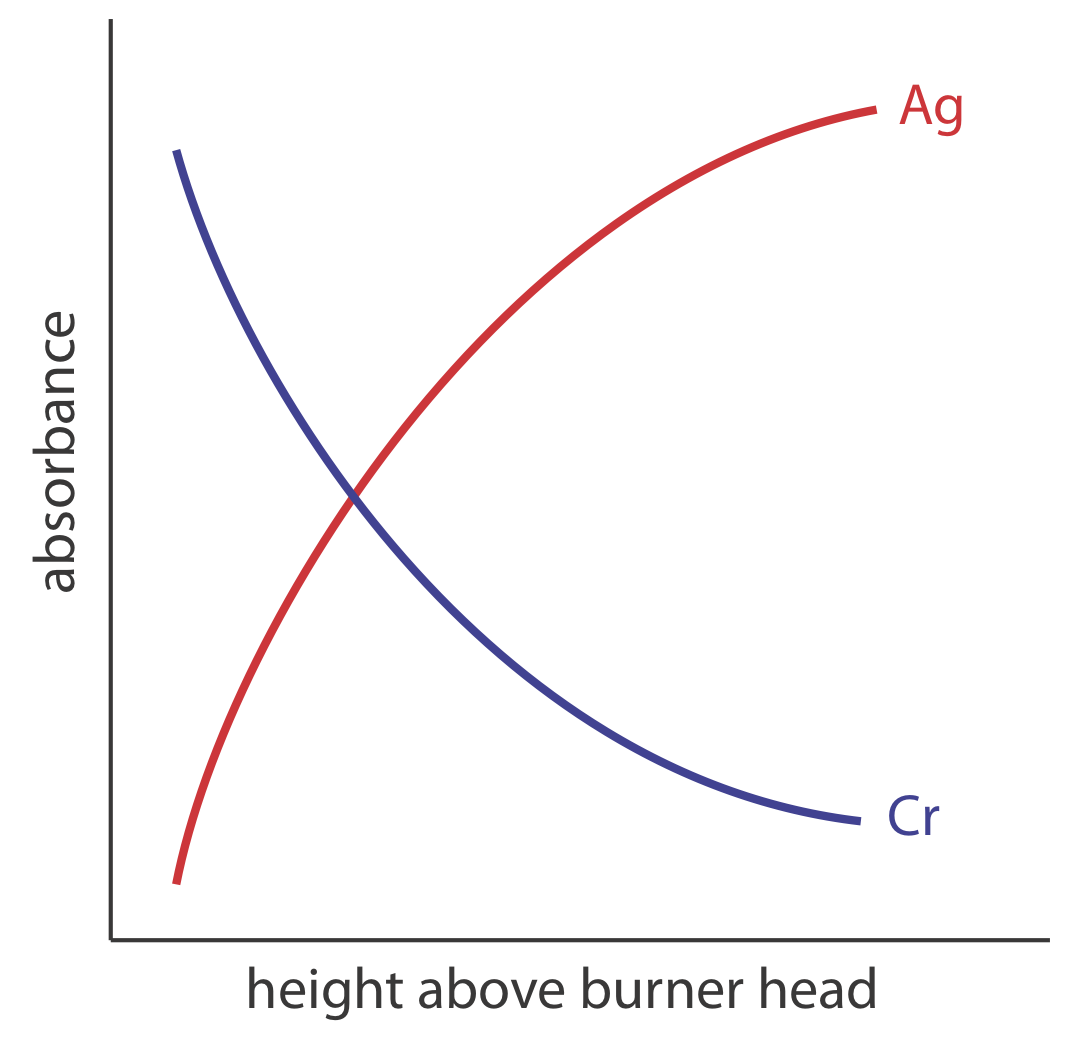
Полум'я. Температура полум'я, яка впливає на ефективність розпилення, залежить від суміші паливно-окислювача, кілька прикладів якої наведено в таблиці Template:index. З них, повітря - ацетилен і оксид азоту - ацетилен полум'я є найбільш популярними. Зазвичай паливо та окислювач змішуються приблизно в стехіометричному співвідношенні; однак для легко окислюваних аналітів може знадобитися багата паливом суміш.
| паливо | окислювач | Діапазон температур (o C) |
|---|---|---|
| природний газ | повітря | 1700—1900 |
| водню | повітря | 2000—2100 |
| ацетилену | повітря | 2100—2400 |
| ацетилену | закис азоту | 2600—2800 |
| ацетилену | кисень | 3050—3150 |
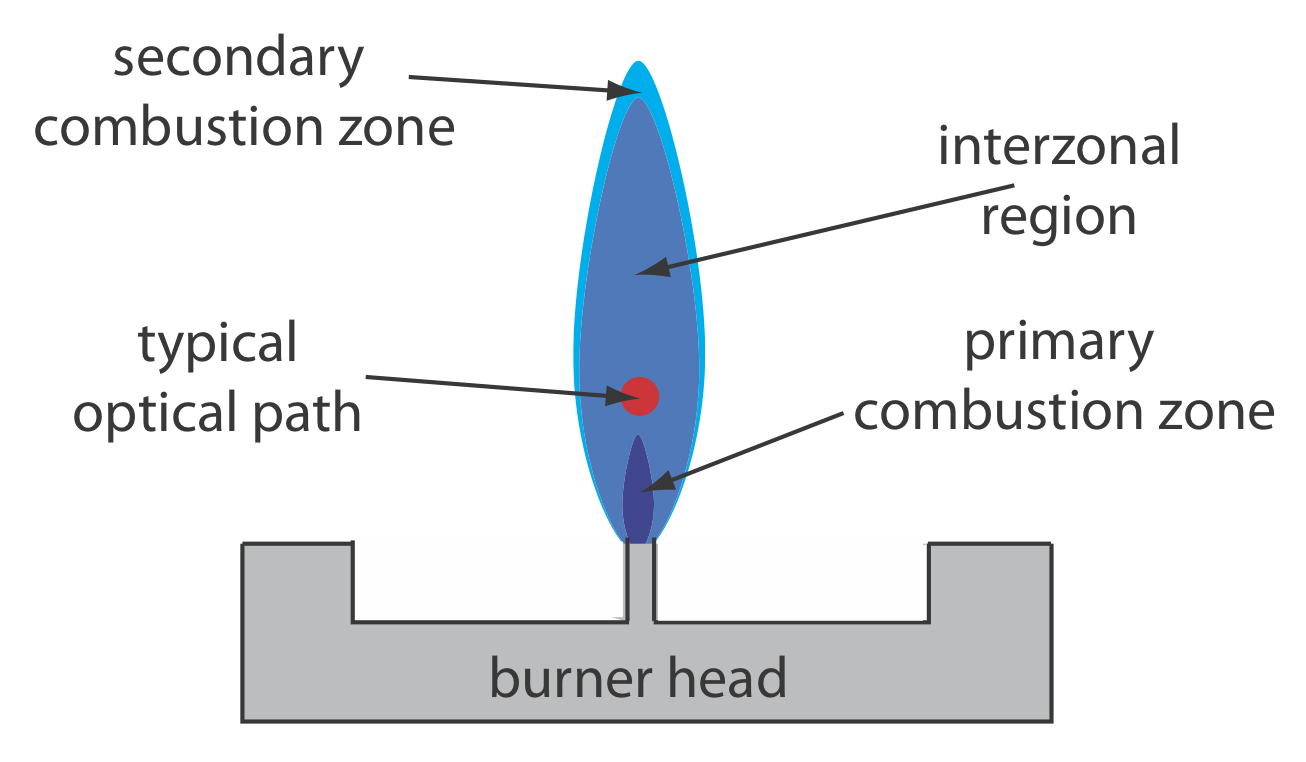
Рисунок Template:index показує поперечний переріз через полум'я, дивлячись вниз по оптичному шляху джерела випромінювання. Первинна зона горіння зазвичай багата продуктами згоряння газу, які випромінюють випромінювання, обмеження корисного для атомного поглинання. Інтерзональна область, як правило, багата вільними атомами і забезпечує найкраще місце для вимірювання атомного поглинання. Найгарячіша частина полум'я зазвичай знаходиться на 2-3 см вище зони первинного горіння. Коли атоми наближаються до вторинної зони горіння полум'я, зниження температури дозволяє формувати стабільні молекулярні види.
Введення зразка. Найпоширенішим засобом для введення проби в розпилювач полум'я є безперервна аспірація, при якій зразок протікає через пальник, поки ми контролюємо поглинання. Безперервна аспірація є інтенсивною пробою, зазвичай вимагає від 2—5 мл зразка.
Мікродискретизація полум'я дозволяє ввести дискретний зразок фіксованого об'єму, і корисний, якщо у нас обмежена кількість проби або коли матриця зразка несумісна з розпилювачем полум'я. Наприклад, безперервне аспірація зразка, який має високу концентрацію розчинених твердих речовин - морська вода, наприклад, приходить на розум - може накопичувати тверду де-позицію на голівці пальника, яка перешкоджає полум'я і знижує поглинання. Мікровідбір полум'я здійснюється за допомогою мікропіпета для розміщення 50-250 мкл зразка в тефлонову воронку, підключену до розпилювача, або зануренням трубки небулайзера в зразок на короткий час. Відбір проб, як правило, здійснюється за допомогою автоматичного пробовідбірника. Сигналом для мікродискретизації полум'я є перехідний пік, висота або площа якого пропорційні кількості аналіту, який вводиться.
Переваги та недоліки розпилення полум'я. Основною перевагою розпилення полум'я є відтворюваність, з якою зразок вводиться в спектрофотометр; істотним недоліком є те, що ефективність розпилення досить низька. Є дві причини поганої ефективності розпилення. По-перше, більшість аерозольних крапель, що утворюються при розпилюванні, занадто великі, щоб їх можна було переносити до полум'я газами згоряння. Отже, до 95% зразка ніколи не досягає полум'я, що є причиною відходів лінії, показаної внизу розпилювальної камери на малюнку Template:index. Друга причина поганої ефективності розпилення полягає в тому, що великий обсяг газів згоряння значно розріджує пробу. Разом ці внески в ефективність розпилення знижують чутливість, оскільки концентрація аналіта в полум'ї може бути фактором\(2.5 \times 10^{-6}\) меншим, ніж у розчині [Інгл, Дж.; Крауч, С.Р. Спектрохімічний аналіз, Прентіс-Холл: Енглвуд Кліффс, штат Нью-Джерсі, 1988; p. 275].
Електротермічні атомайзери
Значне поліпшення чутливості досягається за рахунок використання резистивного нагрівання графітової трубки замість полум'я. Типовий електротермічний розпилювач, також відомий як графітова піч, складається з циліндричної графітової трубки довжиною приблизно 1-3 см і діаметром 3—8 мм. Як показано на малюнку Template:index, графітова трубка розміщена в герметичному вузлі, який має оптично прозоре вікно на кожному кінці. Через піч пропускається безперервний потік інертного газу, який захищає графітову трубку від окислення і видаляє газоподібні продукти, що утворюються при розпиленні. Блок живлення використовується для пропускання струму через графітову трубку, в результаті чого відбувається резистивний нагрів.
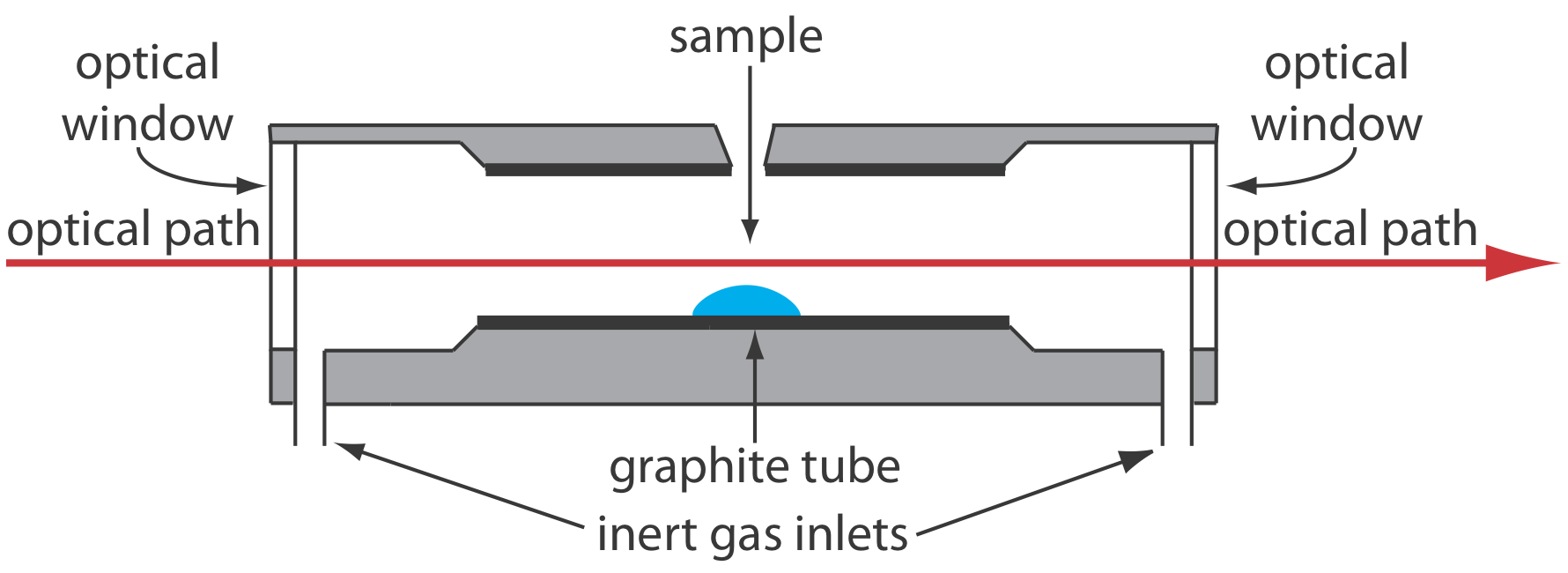
Зразки розміром від 5 до 50 мкл вводяться в графітову трубку через невеликий отвір у верхній частині трубки. Розпилення досягається в три етапи. На першому етапі зразок сушать до твердого залишку за допомогою струму, який підвищує температуру графітової трубки приблизно до 110 o С. На другому етапі, який називається озолом, температуру підвищують до 350—1200 o С. При цих температурах органічного матеріалу в зразок перетворюється в СО 2 і Н 2 О, а летючі неорганічні матеріали випаровуються. Ці гази видаляються потоком інертного газу. На заключній стадії зразок розпилюється шляхом швидкого підвищення температури між 2000-3000 o С. Результатом є перехідний пік поглинання, висота або площа якого пропорційні абсолютній кількості аналіту, що вводиться в графітову трубку. Разом три етапи займають приблизно 45—90 с, причому більшу частину цього часу використовується для сушіння та золи зразка.
Електротермічне розпилення забезпечує значне поліпшення чутливості шляхом уловлювання газоподібного аналіту в малому об'ємі всередині графітової трубки. Концентрація аналіта в результуючій паровій фазі стільки ж\(1000 \times\) більше, ніж при розпиленні полум'я [Parsons, ML.; Major, S.; Forster, A.R. Appl. Спектрозний. 1983, 37, 411—418]. Це поліпшення чутливості і, як наслідок, поліпшення меж виявлення - компенсується значним зниженням точності. На ефективність розпилення сильно впливає контакт зразка з графітовою трубкою, яку важко відтворити.
Різні методи розпилення
Кілька елементів розпилюються за допомогою хімічної реакції для отримання летючого продукту. Такі елементи, як As, Se, Sb, Bi, Ge, Sn, Te і Pb, наприклад, утворюють летючі гідриди, коли вони реагують з NaBH 4 в присутності кислоти. Інертний газ переносить летючий гідрид або до полум'я, або до нагрітої кварцової спостережної трубки, розташованої в оптичному тракті. Ртуть визначається методом холодної пари, при якому вона зводиться до елементарної ртуті з SnCl 2. Летючий Hg переноситься інертним газом до ненагрітої спостережної трубки, розташованої в оптичному шляху приладу.
Кількісні програми
Атомне поглинання широко використовується для аналізу мікрометалів у різних матрицях зразків. Використовуючи Zn як приклад, існують стандартні методи атомного поглинання для його визначення у зразках, таких різноманітних, як вода та стічні води, повітря, кров, сеча, м'язова тканина, волосся, молоко, сухі сніданки, шампуні, сплави, промислові ванни для покриття, бензин, олія, відкладення та гірські породи.
Розробка методу кількісного атомного поглинання вимагає кількох міркувань, включаючи вибір методу атомізації, вибір довжини хвилі та ширини щілини, підготовку зразка до аналізу, мінімізацію спектральних та хімічних перешкод та вибір методу стандартизації. Кожна з цих тем розглядається в цьому розділі.
Розробка кількісного методу
Полум'я або електротермічне розпилення? Найважливішим фактором при виборі методу розпилення є концентрація аналіта. Через його більшу чутливість потрібно менше аналіту для досягнення заданого поглинання при використанні електротермічного розпилення. Таблиця Template:index, в якій порівнюється кількість аналіту, необхідного для досягнення поглинання 0,20 при використанні розпилення полум'я та електротермічного розпилення, корисна при виборі методу розпилення. Наприклад, розпилення полум'ям є методом вибору, якщо наші зразки містять 1-10 мг Zn 2 + /L, але електротермічне розпилення є найкращим вибором для зразків, які містять 1-10 мкг Zn 2 + /L.
| елемент | розпилення полум'я | електротермічне розпилення |
|---|---|---|
| Ag | 1.5 | 0,0035 |
| Аль | 40 | 0,015 |
| Як | 40 | 0,050 |
| Ca | 0.8 | 0,003 |
| Cd | 0.6 | 0,001 |
| Co | 2.5 | 0,021 |
| Cr | 2.5 | 0,0075 |
| Cu | 1.5 | 0,012 |
| Fe | 2.5 | 0,006 |
| Hg | 70 | 0,52 |
| Мг | 0,15 | 0.00075 |
| Мн | 1 | 0,003 |
| Na | 0.3 | 0.00023 |
| Ni | 2 | 0,024 |
| Пб | 5 | 0,080 |
| Пт | 70 | 0,29 |
| Сн | 50 | 0,023 |
| Zn | 0.3 | 0,00071 |
|
Джерело: Варіан Кулінарна книга, Програмне забезпечення SpectraAA Версія 4.00 Pro. Як: 10 мг/л шляхом випаровування гідридів; Hg: 11,5 мг/л холодною парою; і Sn:18 мг/л шляхом випаровування гідридів |
||
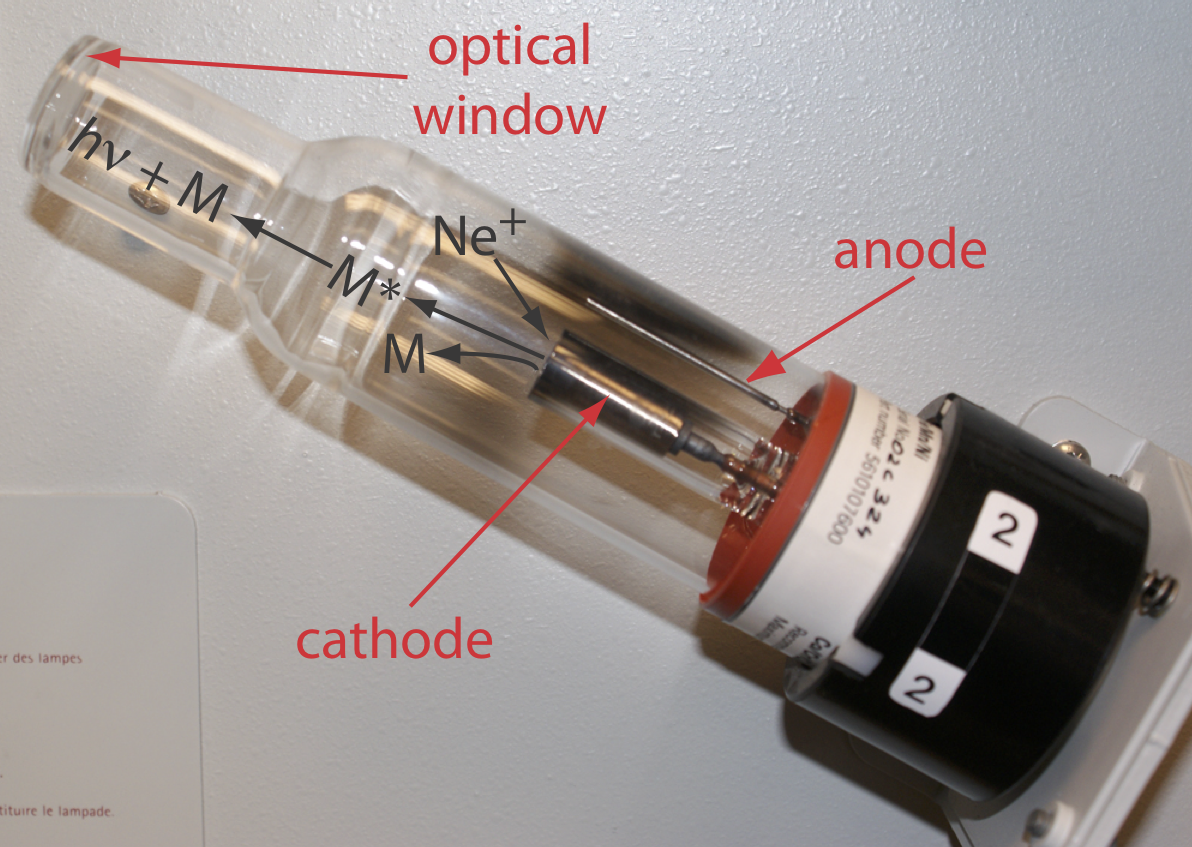
Вибір довжини хвилі та ширини щілини. Джерелом атомного поглинання є порожниста катодна лампа, яка складається з катода і анода, укладених в скляну трубку, заповнену низьким тиском інертного газу, такого як Ne або Ar (рис. Template:index). Застосування потенціалу через електроди іонізує газ наповнювача. Позитивно заряджені іони газу стикаються з негативно зарядженим катодом, розпорошуючи атоми з поверхні катода. Деякі з розпилених атомів знаходяться в збудженому стані і випромінюють випромінювання, характерне для металу (ів), з якого виготовлений катод. Створюючи катод з металевого аналіту, порожниста катодна лампа забезпечує емісійні лінії, які відповідають спектру поглинання аналіта.
Оскільки атомні лінії поглинання вузькі, нам потрібно використовувати лінійне джерело замість джерела континууму (порівняйте, наприклад, рис. 10.2.4 з рисунком 10.2.6). Ефективна пропускна здатність при використанні джерела континууму приблизно\(1000 \times\) більша за лінію атомного поглинання; таким чином, P T ≈ P 0,% T ≈ 100, а A ≈ 0. Оскільки лампа з порожнистим катодом є лінійним джерелом, P T та P 0 мають різні значення, що дають% T < 100 та A > 0.
Кожен елемент у лампі з порожнистим катодом забезпечує кілька атомних емісійних ліній, які ми можемо використовувати для атомного поглинання. Зазвичай довжина хвилі, яка забезпечує найкращу чутливість, є тією, яку ми обираємо використовувати, хоча менш чутлива довжина хвилі може бути більш підходящою для зразка, який має більш високу концентрацію аналіту. Для лампи з порожнистим катодом Cr в таблиці Template:index найкраща чутливість отримана за допомогою довжини хвилі 357,9 нм.
| довжина хвилі (нм) | ширина щілини (нм) | мг Ср/л, що дає А = 0,20 | P 0 (відносний) |
|---|---|---|---|
| 357.9 | 0.2 | 2.5 | 40 |
| 425.4 | 0.2 | 12 | 85 |
| 429.0 | 0.5 | 20 | 100 |
| 520.5 | 0.2 | 1500 | 15 |
| 520.8 | 0.2 | 500 | 20 |
Іншим міркуванням є інтенсивність емісійної лінії. Якщо кілька емісійних ліній відповідають нашим вимогам щодо чутливості, ми можемо використовувати лінію викидів з найбільшим відносним P 0, оскільки існує менша невизначеність у вимірюванні P 0 та P T. Наприклад, при аналізі зразка, який становить ≈10 мг Cr/L, перші три довжини хвиль у таблиці Template:index забезпечують відповідну чутливість; довжини хвиль 425,4 нм і 429,0 нм, однак, мають більший P 0 і забезпечать меншу невизначеність у вимірюваному поглинанні.
Спектр випромінювання для лампи з порожнистим катодом включає, крім емісійних ліній аналіту, додаткові емісійні лінії від домішок, присутніх в металевому катоді, і від газового наповнювача. Ці додаткові лінії є потенційним джерелом бродячого випромінювання, яке може призвести до інструментального відхилення від закону Біра. Ширина щілини монохроматора встановлена максимально широкою для поліпшення пропускної здатності випромінювання і досить вузькою для усунення цих джерел бродячого випромінювання.
Підготовка зразка. Полум'я та електротермічне розпилення вимагають, щоб аналіт знаходився в розчині. Тверді зразки вносять в розчин шляхом розчинення у відповідному розчиннику. Якщо зразок не розчинний, його перетравлюють або на плиті, або мікрохвильовою піччю, використовуючи HNO 3, H 2 SO 4 або HClO 4. Крім того, ми можемо витягти аналіт за допомогою екстрактора Сокслета. Рідкі зразки аналізуються безпосередньо або аналіти витягуються, якщо матриця сумісна з методом розпилення. Наприклад, зразок сироватки важко аспірації при використанні розпилення полум'я і може призвести до неприпустимо високого поглинання фону при використанні електротермічного розпилення. Рідинно-рідка екстракція з використанням органічного розчинника та хелатного агента часто використовується для концентрації аналітів. Розбавлені розчини Cd 2 +, Co 2+, Cu 2 +, Fe 3+, Pb 2 +, Ni 2 +, і Zn 2 + концентрують, наприклад, шляхом екстрагування розчином амонію дітіокарбамат піролідину в метил-ізобутилкетоні.
Мінімізація спектральних перешкод. Спектральна інтерференція виникає, коли лінія поглинання аналіта перекривається лінією поглинання інтерферента або смугою. Оскільки вони настільки вузькі, перекриття двох ліній атомного поглинання рідко є проблемою. З іншого боку, широка смуга поглинання молекули або розсіювання випромінювання джерела є потенційно серйозною спектральною перешкодою.
Важливим фактором при використанні полум'я як джерела розпилення є його вплив на вимірювану поглинання. Серед продуктів згоряння є молекулярні види, які демонструють широкі смуги поглинання і частинки, які розсіюють випромінювання від джерела. Якщо нам не вдається компенсувати ці спектральні перешкоди, то інтенсивність випромінювання, що передається менше, ніж очікувалося. Результатом є очевидне збільшення поглинання зразка. На щастя, поглинання і розсіювання випромінювання полум'ям коригуються аналізом заготовки.
Спектральні перешкоди також виникають, коли компоненти матриці зразка, крім аналіту, реагують на утворення молекулярних видів, таких як оксиди та гідроксиди. Отримане поглинання та розсіювання є фоном зразка і може представляти значну проблему, особливо на довжині хвиль нижче 300 нм, де розсіювання випромінювання стає більш важливим. Якщо нам відомий склад матриці зразка, то ми можемо підготувати наші зразки, використовуючи ідентичну матрицю. У цьому випадку поглинання фону однакове як для зразків, так і для стандартів. Крім того, якщо фон обумовлений відомим компонентом матриці, то ми можемо додати цей компонент в надлишку до всіх зразків і стандартів, так що внесок природного інтерференту незначний. Нарешті, багато перешкод, обумовлених матрицею зразка, усуваються збільшенням температури розпилення. Наприклад, перемикання на більш високу температуру полум'я допомагає запобігти утворенню заважають оксидів і гідроксидів.
Якщо ідентичність перешкод матриці невідома, або якщо неможливо відрегулювати умови полум'я або печі для усунення перешкод, то ми повинні знайти інший метод компенсації фонових перешкод. Для компенсації матричних перешкод було розроблено кілька методів, і більшість атомно-абсорбційних спектрофотометрів включають один або кілька з цих методів.
Одним з найпоширеніших методів корекції фону є використання джерела континууму, такого як лампа D 2. Оскільки лампа D 2 є джерелом континууму, поглинання її випромінювання вузькою лінією поглинання аналіту незначне. Тільки фон, таким чином, поглинає випромінювання від лампи D 2. І аналіт, і фон, з іншого боку, поглинають випромінювання порожнистого катода. Віднімання поглинання для лампи D 2 від поглинання для лампи з порожнистим катодом дає виправлене поглинання, яке компенсує фонові перешкоди. Хоча цей метод корекції фону є ефективним, він припускає, що поглинання фону є постійним у діапазоні довжин хвиль, що передаються монохроматором. Якщо це не так, то віднімання двох поглинань занижує або завищує фон.
Розроблено й інші методи фонової корекції, включаючи корекцію фону ефекту Зеемана та корекцію фону Сміта-Хіфтьє, обидва з яких включені в деякі комерційно доступні атомно-абсорбційні спектрофотометри. Зверніться до додаткових ресурсів глави для отримання додаткової інформації.
Мінімізація хімічних перешкод. Кількісний аналіз деяких елементів ускладнюється хімічними перешкодами, що виникають при атомізації. Найбільш поширеними хімічними перешкодами є утворення нелетких сполук, які містять аналіт і іонізація аналіту.
Одним із прикладів утворення нелеткої сполуки є вплив\(\text{PO}_4^{3-}\) або Al 3 + на аналіз атомного поглинання полум'я Ca 2 +. В одному дослідженні, наприклад, додавання 100 ppm Al 3 + до розчину 5 ppm Ca 2 + знизило абсорбцію іона кальцію з 0,50 до 0,14, при додаванні 500 ppm\(\text{PO}_4^{3-}\) до аналогічного розчину Ca 2 + знизило поглинання з 0,50 до 0,38. Ці перешкоди пояснюються утворенням нелетких частинок Ca 3 (PO 4) 2 та оксиду Al—Ca—O [Hosking, JW; Snell, N.B.; Sturman, B.T. J. Chem. Едук. 1977, 54, 128—130].
При використанні розпилення полум'я ми можемо мінімізувати утворення нелетких сполук за рахунок підвищення температури полум'я шляхом зміни співвідношення палива до окислювача або шляхом переходу на іншу комбінацію палива і окислювача. Інший підхід полягає в додаванні до зразка вивільняючого агента або захисного агента. А р е лізинг агент є вид, який реагує переважно з інтерферентом, вивільняючи аналіт під час розпилення. Наприклад, Sr 2 + і La 3 + служать рилізинг-агентами для аналізу Са 2 + в присутності\(\text{PO}_4^{3-}\) або Al 3 +. Додавання 2000 проміле SrCl 2 до Ca 2 +/\(\text{PO}_4^{3-}\)і до сумішей Ca 2 + + /Al 3+, описаних в попередньому пункті, збільшило поглинання до 0,48. Захисний агент реагує з аналітом, утворюючи стабільний летючий комплекс. Додавання 1% w/w EDTA до\(\text{PO}_4^{3-}\) розчину Ca 2 +/, описаного в попередньому пункті, збільшило поглинання до 0,52.
Іонізаційна перешкода виникає, коли теплової енергії від полум'я або електротермічного розпилювача достатньо для іонізації аналіту.
\[\mathrm{M}(s)\rightleftharpoons \ \mathrm{M}^{+}(a q)+e^{-} \label{10.1}\]
де M - аналіт. Оскільки спектри поглинання для M і M + різні, положення рівноваги в реакції\ ref {10.1} впливає на поглинання на довжині хвиль, де M поглинає. Щоб обмежити іонізацію, ми додаємо високу концентрацію придушувача іонізації, який є видом, який іонізується легше, ніж аналіт. Якщо концентрація придушувача іонізації достатня, то підвищена концентрація електронів у полум'ї штовхає реакцію\ ref {10.1} вліво, перешкоджаючи іонізації аналіта. Калій і цезій часто використовуються як придушувач іонізації через їх низьку енергію іонізації.
Стандартизація методу. Оскільки закон Біра також застосовується до атомного поглинання, ми можемо очікувати, що криві калібрування атомної абсорбції будуть лінійними. Однак на практиці більшість калібрувальних кривих атомного поглинання є нелінійними або лінійними в обмеженому діапазоні концентрацій. Нелінійність атомного поглинання є наслідком інструментальних обмежень, включаючи блукаюче випромінювання від лампи з порожнистим катодом і зміна молярної поглинання по лінії поглинання. Точна кількісна робота, отже, вимагає відповідного засобу для обчислення калібрувальної кривої з набору стандартів.
Коли це можливо, кількісний аналіз найкраще проводити з використанням зовнішніх стандартів. На жаль, матричні перешкоди є частою проблемою, особливо при використанні електротермічного розпилення. З цієї причини часто використовується метод стандартних доповнень. Однак одним з обмежень цього методу стандартизації є вимога лінійної залежності між поглинанням та концентрацією.
Більшість приладів включають кілька різних алгоритмів обчислення калібрувальної кривої. Наприклад, прилад у моїй лабораторії включає п'ять алгоритмів. Три алгоритми підходять до даних поглинання за допомогою лінійних, квадратичних або кубічних поліноміальних функцій концентрації аналіта. Він також включає два алгоритми, які відповідають концентрації стандартів квадратичним функціям поглинання.
Представницький метод 10.4.1: Визначення Cu та Zn у зразках тканин
Найкращий спосіб оцінити теоретичні та практичні деталі, розглянуті в цьому розділі, - це уважно вивчити типовий аналітичний метод. Хоча кожен метод унікальний, наступний опис визначення Cu і Zn в біологічних тканині дає повчальний приклад типової процедури. Опис тут базується на Бхаттачарії, С.К.; Гудвін, Т. Г.; Кроуфорд, А.Дж. анал. Летт. 1984, 17, 1567—1593, і Кроуфорд, А.Дж.; Бхаттачарія, С.К. Варіан Інструменти на роботі, номер AA—46, квітень 1985.
Опис методу.
Мідь і цинк виділяють із зразків тканин шляхом перетравлення зразка з HNO 3 після першого видалення будь-якої жирової клітковини. Концентрацію міді і цинку в супернатанті визначають атомним поглинанням за допомогою повітряно-ацетиленового полум'я.
Порядок дій.
Зразки тканин отримують біопсією м'язової голки і сушать протягом 24-30 ч при 105 о С для видалення всіх слідів вологи. Жирова клітковина в висушеному зразку видаляється шляхом екстракції протягом ночі безводним ефіром. Після видалення ефіру зразок висушують, щоб отримати знежирену масу сухої тканини (FFDT). Пробу перетравлюють при 68 o С протягом 20-24 ч з використанням 3 мл 0,75 М HNO 3. Після центрифугування при 2500 об/хв протягом 10 хвилин супернатант переносять в об'ємну колбу об'ємом 5 мл. Травлення повторюють ще два рази, по 2-4 години кожен, використовуючи 0,9-мл аліквоти 0,75 М HNO 3. Ці супернатанти додаються в об'ємну колбу об'ємом 5 мл, яка розбавляється до об'єму 0,75 М HNO 3. Концентрації Cu і Zn в розведеному супернатанті визначають методом атомно-абсорбційної спектроскопії полум'я з використанням повітряно-ацетиленового полум'я та зовнішніх стандартів. Мідь аналізується на довжині хвилі 324,8 нм при ширині щілини 0,5 нм, а цинк аналізується при 213,9 нм при ширині щілини 1,0 нм. Корекція фону за допомогою лампи D 2 необхідна для цинку. Результати повідомляються у вигляді мкг Cu або Zn на грам FFDT.
Питання.
1. Опишіть відповідну матрицю для зовнішніх стандартів і для бланка?
Матриця для стандартів і бланк повинні збігатися з матрицею зразків; таким чином, відповідна матриця дорівнює 0,75 М HNO 3. Будь-які перешкоди від інших компонентів матриці зразка мінімізуються за допомогою фонової корекції.
2. Чому фонова корекція необхідна для аналізу Zn, але не для аналізу Cu?
Фонова корекція компенсує поглинання та розсіювання фону за рахунок перешкод у вибірці. Такі перешкоди найбільш серйозні при використанні довжини хвилі менше 300 нм. Це стосується Zn, але не для Cu.
3. Лампа з порожнистим катодом Cu має кілька емісійних ліній, властивості яких наведені в наступній таблиці. Поясніть, чому цей метод використовує лінію на 324,8 нм.
| довжина хвилі (нм) | ширина щілини (нм) | мг Ку/л для А = 0,20 | P 0 (відносний) |
|---|---|---|---|
| 217,9 | 0.2 | 15 | 3 |
| 218.2 | 0.2 | 15 | 3 |
| 222.6 | 0.2 | 60 | 5 |
| 244.2 | 0.2 | 400 | 15 |
| 249.2 | 0.5 | 200 | 24 |
| 324,8 | 0.5 | 1.5 | 100 |
| 327,4 | 0.5 | 3 | 87 |
При 1,5 мг Cu/L, що дає поглинання 0,20, емісійна лінія при 324,8 нм має найкращу чутливість. Крім того, це найбільш інтенсивна емісійна лінія, яка зменшує невизначеність вимірюваного поглинання.
Для оцінки методу, описаного в репрезентативному методі 10.4.1, підготовлено та проаналізовано серію зовнішнього стандарту, надаючи результати, показані тут [Кроуфорд, А.Дж.; Bhattacharya, SK «Мікроаналіз міді та цинку в зразках тканин розміром з біопсію методом атомної абсорбційної спектроскопії з використанням Стехіометричний повітряно-ацетиленовий полум'я,» Varian Instruments at Work, номер AA—46, квітень 1985].
| мкг Cu/мл | поглинання | мкг Cu/мл | поглинання |
|---|---|---|---|
| 0.000 | 0.000 | 0,500 | 0,033 |
| 0.100 | 0,006 | 0.600 | 0.039 |
| 0,200 | 0,013 | 0,700 | 0.046 |
| 0,300 | 0,020 | 1.00 | 0.066 |
| 0,400 | 0.026 |
Для оцінки точності методу використовується стандартний еталонний матеріал бичачої печінки. Після сушіння та вилучення зразка зразок тканини FFDT 11,23 мг дає поглинання 0,023. Повідомте про кількість міді в зразку як мкг Cu/g FFDT.
Рішення
Лінійна регресія поглинання в порівнянні з концентрацією Cu в стандартах дає калібрувальну криву, показану нижче, і наступне калібрувальне рівняння.
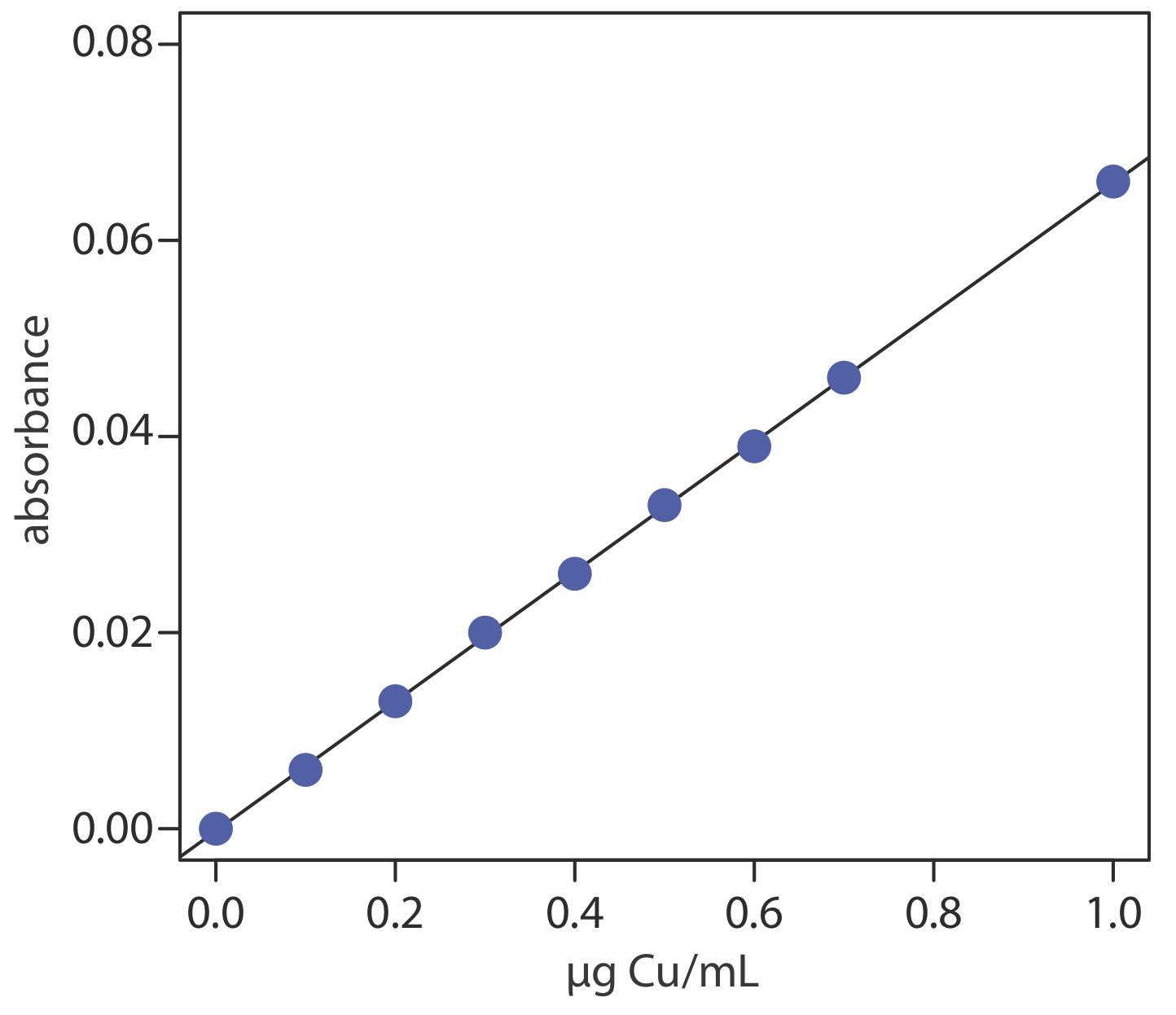
\[A=-0.0002+0.0661 \times \frac{\mu \mathrm{g} \ \mathrm{Cu}}{\mathrm{mL}} \nonumber\]
Підстановка поглинання зразка в калібрувальне рівняння дає концентрацію міді 0,351 мкг/мл. Отже, концентрація міді в зразку тканини становить
\[\frac { \frac{0.351 \mu \mathrm{g} \ \mathrm{Cu}}{\mathrm{mL}} \times 5.000 \ \mathrm{mL}} {0.01123 \text{ g sample}}=156 \ \mu \mathrm{g} \ \mathrm{Cu} / \mathrm{g} \ \mathrm{FDT} \nonumber\]
Оцінка атомно-абсорбційної спектроскопії
Масштаб операції
Атомно-абсорбційна спектроскопія ідеально підходить для аналізу слідів та ультратрасових аналітів, особливо при використанні електротермічного розпилення. Для незначних і основних аналітів пробу розводять перед аналізом. Більшість аналізів використовують макрос або мезозразок. Однак невелика потреба в об'ємі для електротермічного розпилення або мікровідбору проб полум'я робить практичним аналіз мікро- та ультрамікро зразків.
Точність
Якщо спектральні та хімічні перешкоди зведені до мінімуму, зазвичай досягається точність 0,5— 5%. Коли калібрувальна крива нелінійна, точність покращується за допомогою пари стандартів, поглинання яких тісно обмежують поглинання зразка, і припускаючи, що зміна поглинання є лінійною в цьому обмеженому діапазоні концентрацій. Визначені похибки електротермічного розпилення часто більше, ніж ті, що отримані при розпиленні полум'я через більш серйозні перешкоди матриці.
Точність
Для поглинання, що перевищує 0,1-0,2, відносне стандартне відхилення атомного поглинання становить 0,3— 1% для розпилення полум'я і 1-5% для електротермічного розпилення. Основним обмеженням є невизначеність концентрації вільних атомів аналіту, що виникає внаслідок варіацій швидкості аспірації, розпилення та розпилення для розпилювача полум'я, а також послідовність ін'єкційних зразків для електротермічного розпилення.
Чутливість
На чутливість аналізу атомного поглинання полум'я впливає склад полум'я і положення в полум'ї, з якого ми контролюємо поглинання. Зазвичай чутливість аналізу оптимізується шляхом аспірації стандартного розчину аналіту та регулювання співвідношення палива до окислювача, швидкості потоку небулайзера та висоти пальника, щоб забезпечити найбільше поглинання. При електротермічному розпиленні на чутливість впливають стадії сушіння та золи, які передують розпиленню. Температура і час на кожному етапі оптимізовані для кожного типу зразка.
На чутливість також впливає матриця зразка. Ми вже відзначали, наприклад, що чутливість знижується хімічним втручанням. Підвищення чутливості може бути реалізовано шляхом додавання в розчин низькомолекулярного спирту, ефіру або кетону, або за допомогою органічного розчинника.
Вибірковість
Завдяки вузькій ширині ліній поглинання атомне поглинання забезпечує чудову селективність. Атомна абсорбція використовується для аналізу понад 60 елементів при концентраціях при або нижче рівня мкг/л.
Час, вартість та обладнання
Час аналізу при використанні полум'яного розпилення короткий, при використанні повністю автоматизованої системи пропускна здатність зразка становить 250-350 визначень на годину. Електротермічне розпилення вимагає значно більше часу на аналіз, з максимальною пропускною здатністю зразків 20—30 визначень на годину. Вартість нового приладу коливається від 10 000 до 50 000 доларів за розпилення полум'я та від 18 000 до 70 000 доларів для електротермічного розпилення. Більш дорогі прилади в кожному ціновому діапазоні включають двопроменеву оптику, автоматичні пробовідбірники, і їх можна запрограмувати на багатоелементний аналіз, дозволяючи автоматично змінювати довжину хвилі та порожнисту катодну лампу.