1.5: Калібрування інструментальних методів
- Page ID
- 26980

Щоб стандартизувати аналітичний метод, ми також повинні визначити чутливість аналіта, k A, у наступному рівнянні
\[S_{total} = k_A C_A + S_{blank} \label{s_total} \]
де\(S_{total}\) - вимірюваний сигнал,\(C_A\) - концентрація аналіта, і\(S_{reag}\) сигнал при відсутності аналіта. В принципі, можна вивести значення k A для будь-якого аналітичного методу, якщо зрозуміти повністю всі хімічні реакції і фізичні процеси, що відповідають за сигнал. На жаль, такі розрахунки нездійсненні, якщо бракує достатньо розвиненої теоретичної моделі фізичних процесів або якщо хімічна реакція свідчить про неідеальну поведінку. У таких ситуаціях ми повинні визначити значення k A шляхом аналізу одного або декількох стандартних рішень, кожен з яких містить відому кількість аналіту. У цьому розділі розглянуто кілька підходів до визначення величини k A. Для простоти ми припускаємо, що\(S_{blank}\) враховується належним порожнім реагентом, що дозволяє нам замінити S total in сигналом аналіта, S A.
\[S_A = k_A C_A \label{sa} \]
Одноточкова проти множинної стандартизації
Найпростішим способом визначення значення k A в Equation\ ref {sa} є використання одноточкової стандартизації, в якій ми вимірюємо сигнал для стандарту S std, який містить відому концентрацію аналіту, C стд. Підставляємо ці значення в Equation\ ref {sa} і переставляємо
\[k_A = \frac {S_{std}} {C_{std}} \label{ka} \]
щоб дати нам значення для k A. Визначивши k A, ми можемо обчислити концентрацію аналіту в зразку шляхом вимірювання його сигналу, S samp, і обчислити C A як
\[C_A = \frac {S_{samp}} {k_A} \label{ca} \]
Одноточкова стандартизація є найменш бажаним методом стандартизації методу. Цьому є дві причини. По-перше, будь-яка похибка в нашому визначенні k A переноситься в наш розрахунок C A. По-друге, наше експериментальне значення для k A засноване на одній концентрації аналіту. Щоб розширити це значення k A до інших концентрацій аналіту, потрібно припустити лінійну залежність між сигналом і концентрацією аналіта, припущення, яке часто не відповідає дійсності [Кардон, М.Дж.; Palmero, P J.; Sybrandt, LB Anal. Хім. 1980, 52, 1187—1191]. На малюнку\(\PageIndex{1}\) показано, як припущення постійної величини k A призводить до визначеної помилки в C A, якщо k A стає меншим при більш високих концентраціях аналіту. Незважаючи на ці обмеження, одноточкові стандартизації знаходять звичайне використання, коли очікуваний діапазон концентрацій аналіта невеликий. У цих умовах часто можна з упевненістю припустити, що k A є постійним (хоча ви повинні перевірити це припущення експериментально). Так відбувається, наприклад, в клінічних лабораторіях, де багато автоматизованих аналізаторів використовують лише єдиний стандарт.
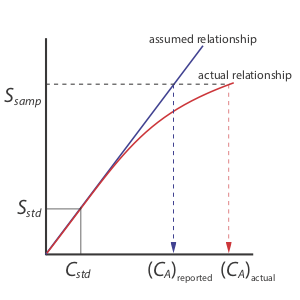
Кращим способом стандартизації методу є підготовка ряду стандартів, кожен з яких містить різну концентрацію аналіту. Стандарти вибираються такі, що вони відповідають очікуваному діапазону концентрації аналіта. Багатоточкова стандартизація повинна включати щонайменше три стандарти, хоча більш кращими є. Графік S std проти C std називається калібрувальною кривою. Точна стандартизація, або залежність калібрування, визначається відповідним алгоритмом підгонки кривої.
Лінійна регресія, яка також відома як метод найменших квадратів, є одним з таких алгоритмів. Його використання розглянуто в Додатку 1.
Є дві переваги багатоточкової стандартизації. По-перше, хоча детермінантна помилка в одному стандарті вносить детермінантну похибку, її вплив зводиться до мінімуму іншими стандартами. По-друге, оскільки ми вимірюємо сигнал для декількох концентрацій аналіту, ми більше не повинні вважати, що k A не залежить від концентрації аналіта. Натомість ми можемо побудувати калібрувальну криву, подібну до «фактичного співвідношення» на малюнку\(\PageIndex{1}\).
Зовнішні стандарти
Найбільш поширений метод стандартизації використовує один або кілька зовнішніх стандартів, кожен з яких містить відому концентрацію аналіту. Ми називаємо ці стандарти «зовнішніми», оскільки вони готуються і аналізуються окремо від зразків.
Додавання прикметника «зовнішній» до іменника «стандарт» може здатися вам дивним у цей момент, оскільки здається розумним припустити, що стандарти та зразки аналізуються окремо. Однак, як ми скоро дізнаємося, ми можемо додати стандарти до наших зразків і аналізувати обидва одночасно.
Одномісний зовнішній стандарт
За допомогою єдиного зовнішнього стандарту визначаємо k A за допомогою eEquation\ ref {ka}, а потім обчислюємо концентрацію аналіту C A, використовуючи Equation\ ref {ca}.
Спектрофотометричний метод кількісного аналізу Pb 2+ в крові дає S std 0,474 для єдиного стандарту, для якого концентрація свинцю становить 1,75 ppb. Яка концентрація Pb 2 + в зразку крові, для якої S самп дорівнює 0,361?
Рішення
Рівняння\ ref {ka} дозволяє обчислити значення k A, використовуючи дані для єдиного зовнішнього стандарту.
\[k_A = \frac {S_{std}} {C_{std}} = \frac {0.474} {1.75 \text{ ppb}} = 0.2709 \text{ ppb}^{-1} \nonumber \]
Визначивши значення k А, обчислюємо концентрацію Pb 2 + в пробі крові, розраховану за допомогою Equation\ ref {ca}.
\[C_A = \frac {S_{samp}} {k_A} = \frac {0.361} {0.2709 \text{ ppb}^{-1}} = 1.33 \text{ ppb} \nonumber \]
Кілька зовнішніх стандартів
\(\PageIndex{2}\)На малюнку показана типова багатоточкова зовнішня стандартизація. Об'ємна колба зліва містить заготовку реагенту, а решта об'ємних колб містять зростаючі концентрації Cu 2 +. Нижче наведені об'ємні колби - отримана калібрувальна крива. Оскільки це найпоширеніший метод стандартизації, отримане співвідношення називається нормальною калібрувальною кривою.

Коли калібрувальна крива є прямою, як це на малюнку\(\PageIndex{2}\), нахил лінії дає значення k A. Це найбільш бажана ситуація, оскільки чутливість методу залишається постійною у всьому діапазоні концентрацій аналіта. Коли калібрувальна крива не є прямолінійною, чутливість методу є функцією концентрації аналіта. Наприклад\(\PageIndex{1}\), на малюнку значення k A є найбільшим, коли концентрація аналіту мала, і вона постійно зменшується для більш високих концентрацій аналіту. Значення k A в будь-якій точці вздовж калібрувальної кривої на малюнку\(\PageIndex{1}\) є нахилом у цій точці. У будь-якому випадку калібрувальна крива дозволяє пов'язати S samp з концентрацією аналіта.
Другий спектрофотометричний метод кількісного аналізу Pb 2+ в крові має нормальну калібрувальну криву, для якої
\[S_{std} = (0.296 \text{ ppb}^{-1} \times C_{std}) + 0.003 \nonumber \]
Яка концентрація Pb 2 + в зразку крові, якщо S самп дорівнює 0,397?
Рішення
Для визначення концентрації Pb 2 + в зразку крові замінюємо S std в калібрувальному рівнянні на S samp і вирішуємо для C A .
\[C_A = \frac {S_{samp} - 0.003} {0.296 \text{ ppb}^{-1}} = \frac {0.397 - 0.003} {0.296 \text{ ppb}^{-1}} = 1.33 \text{ ppb} \nonumber \]
Варто зазначити, що рівняння калібрування в цій задачі включає додатковий член, який не відображається в Equation\ ref {ca}. В ідеалі ми очікуємо, що наша калібрувальна крива матиме сигнал нуля, коли C A дорівнює нулю. Це мета використання заготовки реагенту для корекції вимірюваного сигналу. Додатковий член +0,003 в нашому калібрувальному рівнянні є результатом невизначеності вимірювання сигналу для заготовки реагенту та стандартів.
Зовнішня стандартизація дозволяє аналізувати серію зразків за допомогою однієї калібрувальної кривої. Це важлива перевага, коли у нас є багато зразків для аналізу. Не дивно, що багато найбільш поширених кількісних аналітичних методів використовують зовнішню стандартизацію.
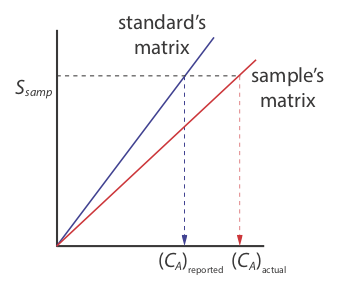
Однак існує серйозне обмеження зовнішньої стандартизації. Коли ми визначаємо значення k A за допомогою Equation\ ref {ka}, аналіт присутній у матриці зовнішнього стандарту, яка зазвичай є набагато простішою матрицею, ніж матриця наших зразків. Коли ми використовуємо зовнішню стандартизацію, ми припускаємо, що матриця не впливає на значення k A. Якщо це не так, то вводимо в наш аналіз пропорційну детермінантну похибку. Це не так на малюнку\(\PageIndex{3}\), наприклад, де ми показуємо калібрувальні криві для аналіту в матриці зразка та в матриці стандарту. У цьому випадку використання калібрувальної кривої для зовнішніх стандартів призводить до негативної детермінантної помилки в повідомленій концентрації аналіту. Якщо ми очікуємо, що матричні ефекти важливі, то ми намагаємося відповідати матриці стандарту з матрицею зразка, процесу, відомого як відповідність матриці. Якщо ми не впевнені в матриці зразка, то ми повинні показати, що матричні ефекти незначні або використовувати альтернативний метод стандартизації. Обидва підходи розглядаються в наступному розділі.
Матриця для зовнішніх стандартів на малюнку\(\PageIndex{2}\), наприклад, - це розведений аміак. Оскільки\(\ce{Cu(NH3)4^{2+}}\) комплекс поглинає сильніше, ніж Cu 2 +, додавання аміаку збільшує величину сигналу. Якщо нам не вдасться додати таку ж кількість аміаку в наші зразки, то ми внесемо в наш аналіз пропорційну детермінантну похибку.
Стандартні доповнення
Уникнути ускладнення узгодження матриці стандартів з матрицею зразка можна, якщо провести стандартизацію в вибірці. Це відомо як метод стандартних доповнень.
Одномісний стандарт з доповненням
Найпростіший варіант стандартного доповнення показаний на рис\(\PageIndex{4}\). Спочатку додаємо порцію зразка, V o, в об'ємну колбу, розводимо її до обсягу, V f, і вимірюємо його сигнал, S samp. Далі ми додаємо другу ідентичну частину зразка до еквівалентної об'ємної колбі разом із шипом, V std, зовнішнього стандарту, концентрація якого становить C std. Після того, як розводимо шиповану пробу до такого ж кінцевого обсягу, заміряємо його сигнал, S шип.
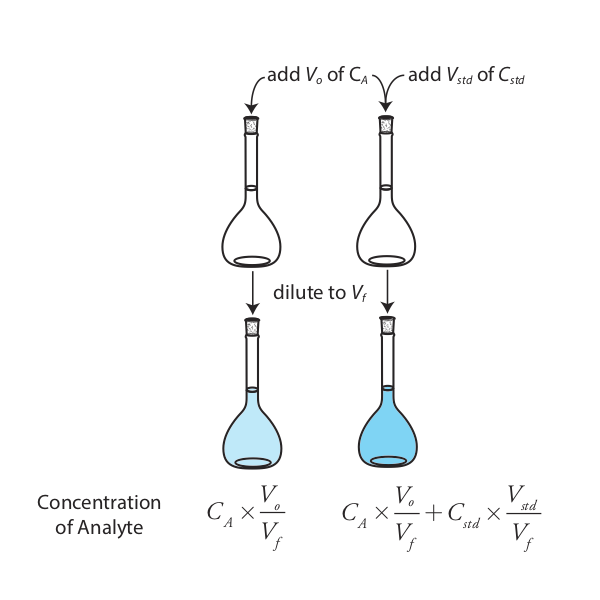
Наступні два рівняння пов'язують S samp і S спайк до концентрації аналіту, C A, у вихідному зразку.
\[S_{samp} = k_A C_A \frac {V_o} {V_f} \label{sa_samp1} \]
\[S_{spike} = k_A \left( C_A \frac {V_o} {V_f} + C_{std} \frac {V_{std}} {V_f} \right) \label{sa_spike1} \]
Поки V std невеликий щодо V o, вплив матриці стандарту на матрицю зразка незначний. За цих умов значення k A однакове в Рівнянні\ ref {sa_samp1} та Рівнянні\ ref {sa_spike1}. Розв'язування обох рівнянь для k A і рівняння дає
\[\frac {S_{samp}} {C_A \frac {V_o} {V_f}} = \frac {S_{spike}} {C_A \frac {V_o} {V_f} + C_{std} \frac {V_{std}} {V_f}} \label{method_one} \]
який ми можемо вирішити для концентрації аналіту, C A, у вихідному зразку.
Третій спектрофотометричний метод кількісного аналізу Pb 2+ в крові дає S samp 0,193 при розведенні 1,00 мл зразка крові до 5,00 мл. Другий зразок крові 1,00 мл шипують 1,00 мл зовнішнього стандарту 1560-ppb Pb 2 + і розбавляють до 5,00 мл, що дає S-шип 0,419. Яка концентрація Pb 2 + в вихідному зразку крові?
Рішення
Ми починаємо з внесення відповідних замін в Equation\ ref {method_one} і рішення для C A. Зверніть увагу, що всі томи повинні бути в однакових одиницях; таким чином, ми спочатку приховаємо V std від 1.00 mL до\(1.00 \times 10^{-3} \text{ mL}\).
\[\frac {0.193} {C_A \frac {1.00 \text{ mL}} {5.00 \text{ mL}}} = \frac {0.419} {C_A \frac {1.00 \text{ mL}} {5.00 \text{ mL}} + 1560 \text{ ppb} \frac {1.00 \times 10^{-3} \text{ mL}} {5.00 \text{ mL}}} \nonumber \]
\[\frac {0.193} {0.200C_A} = \frac {0.419} {0.200C_A + 0.3120 \text{ ppb}} \nonumber \]
\[0.0386C_A + 0.0602 \text{ ppb} = 0.0838 C_A \nonumber \]
\[0.0452 C_A = 0.0602 \text{ ppb} \nonumber \]
\[C_A = 1.33 \text{ ppb} \nonumber \]
Концентрація Pb 2 + в вихідному зразку крові становить 1,33 ppb.
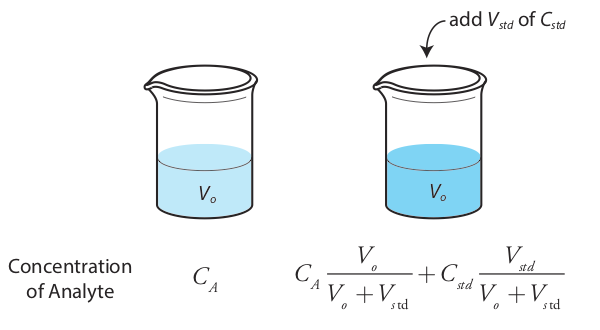
Також можливе додавання стандартного додавання безпосередньо до зразка, вимірюючи сигнал як до, так і після спайка (рис.\(\PageIndex{5}\)). У цьому випадку кінцевим об'ємом після стандартного додавання є V o + V std і Equation\ ref {sa_samp1}, Рівняння\ ref {sa_spike1}, а рівняння\ ref {method_one} стають
\[S_{samp} = k_A C_A \label{sa_samp2} \]
\[S_{spike} = k_A \left( C_A \frac {V_o} {V_o + V_{std}} + C_{std} \frac {V_{std}} {V_o + V_{std}} \right) \label{sa_spike2} \]
\[\frac {S_{samp}} {C_A} = \frac {S_{spike}} {C_A \frac {V_o} {V_o + V_{std}} + C_{std} \frac {V_{std}} {V_o + V_{std}}} \label{method_two} \]
Четвертий спектрофотометричний метод кількісного аналізу Pb 2+ в крові дає S samp 0,712 для зразка крові 5,00 мл. Після спайки зразка крові 5,00 мл зовнішнього стандарту 1560-ppb Pb 2 + вимірюється спайк S 1,546. Яка концентрація Pb 2 + в вихідному зразку крові?
Рішення
\[\frac {0.712} {C_A} = \frac {1.546} {C_A \frac {5.00 \text{ mL}} {5.005 \text{ mL}} + 1560 \text{ ppb} \frac {5.00 \times 10^{-3} \text{ mL}} {5.005 \text{ mL}}} \nonumber \]
\[\frac {0.712} {C_A} = \frac {1.546} {0.9990C_A + 1.558 \text{ ppb}} \nonumber \]
\[0.7113C_A + 1.109 \text{ ppb} = 1.546C_A \nonumber \]
\[C_A = 1.33 \text{ ppb} \nonumber \]
Концентрація Pb 2 + в вихідному зразку крові становить 1,33 ppb.
Кілька стандартних доповнень
Ми можемо адаптувати одноточкове стандартне доповнення до множинного стандартного додавання, підготувавши серію зразків, які містять зростаючі обсяги зовнішнього стандарту. \(\PageIndex{6}\)На малюнку показано два способи побудови стандартної кривої калібрування додавання на основі Equation\ ref {sa_spike1}. На малюнку\(\PageIndex{6}\) а намічаємо S шип проти обсягу шипів, V std. Якщо k A постійна, то калібрувальна крива - пряма. Легко показати, що х -перехоплення еквівалентно — C A V o /C std.
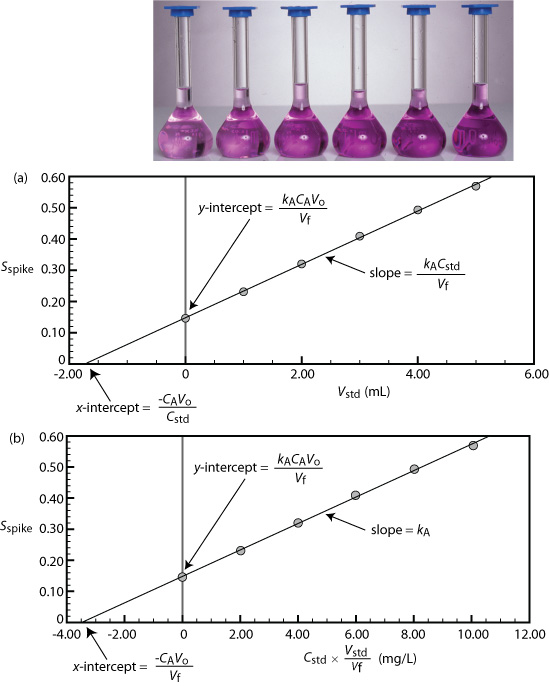
Починаючи з Equation\ ref {sa_spike1} показують, що рівняння на малюнку\(\PageIndex{6}\) a для нахилу, y -перехоплення та x -перехоплення правильні.
Рішення
Ми починаємо з переписування рівняння\ ref {sa_spike1} як
\[S_{spike} = \frac {k_A C_A V_o} {V_f} + \frac {k_A C_{std}} {V_f} \times V_{std} \nonumber \]
який у вигляді рівняння для прямої
\[y = y\text{-intercept} + \text{slope} \times x\text{-intercept} \nonumber \]
де y - S шип, а х - V std. Нахил лінії, отже, дорівнює k A C std/V f, а y -перехоплення дорівнює k A C A В о /В ф. X -intercept - це значення x, коли y дорівнює нулю, або
\[0 = \frac {k_A C_A V_o} {V_f} + \frac {k_A C_{std}} {V_f} \times x\text{-intercept} \nonumber \]
\[x\text{-intercept} = - \frac {k_A C_A V_o / V_f} {K_A C_{std} / V_f} = - \frac {C_A V_o} {C_{std}} \nonumber \]
Оскільки ми знаємо об'єм вихідного зразка, V o, і концентрацію зовнішнього стандарту, C std, ми можемо обчислити концентрації аналіта з x -перехоплення множинних стандартних доповнень.
П'ятий спектрофотометричний метод кількісного аналізу Pb 2+ в крові використовує багатоточкове стандартне додавання на основі Equation\ ref {sa_spike1}. Вихідний зразок крові має об'єм 1,00 мл, а стандарт, який використовується для шипування зразка, має концентрацію 1560 ppb Pb 2 +. Всі зразки були розведені до 5,00 мл перед вимірюванням сигналу. Калібрувальна крива S спайка проти V std має наступне рівняння:
\[S_{spike} = 0.266 + 312 \text{ mL}^{-1} \times V_{std} \nonumber \]
Яка концентрація Pb 2 + в вихідному зразку крові?
Рішення
Щоб знайти x -перехоплення, ми встановимо S шип рівний нулю.
\[S_{spike} = 0.266 + 312 \text{ mL}^{-1} \times V_{std} \nonumber \]
Розв'язуючи для V std, отримаємо значення\(-8.526 \times 10^{-4} \text{ mL}\) для x -перехоплення. Підставляємо значення x -intercept у рівняння з рисунка\(\PageIndex{6}\) a
\[-8.526 \times 10^{-4} \text{ mL} = - \frac {C_A V_o} {C_{std}} = - \frac {C_A \times 1.00 \text{ mL}} {1560 \text{ ppb}} \nonumber \]
а рішення для С А дає концентрацію Pb 2 + в зразку крові як 1,33 ppb.
Оскільки ми будуємо стандартну криву калібрування доповнення в зразку, ми не можемо використовувати калібрувальне рівняння для інших зразків. Кожен зразок, таким чином, вимагає власних стандартних доповнень калібрувальної кривої. Це серйозний недолік, якщо у вас багато зразків. Наприклад, припустимо, вам потрібно проаналізувати 10 зразків за допомогою п'ятиточкової калібрувальної кривої. Для нормальної калібрувальної кривої потрібно проаналізувати всього 15 розчинів (п'ять стандартів і десять зразків). Якщо ви використовуєте метод стандартних доповнень, ви повинні проаналізувати 50 розчинів (кожен з десяти зразків аналізується п'ять разів, один раз перед шипами і після кожного з чотирьох шипів).
Використання стандартного доповнення для ідентифікації матричних ефектів
Ми можемо використовувати метод стандартних доповнень для перевірки зовнішньої стандартизації, коли узгодження матриці неможливо. Спочатку готуємо нормальну калібрувальну криву S std проти C std і визначаємо значення k A з її нахилу. Далі готуємо стандартну доповнення калібрувальної кривої за допомогою Equation\ ref {sa_spike1}, будуючи дані так, як показано на малюнку\(\PageIndex{6}\) b. Ухил цієї стандартної калібрувальної кривої доповнення забезпечує незалежне визначення k A. Якщо між двома значеннями k A немає суттєвої різниці, то ми можемо ігнорувати різницю між матрицею вибірки та матрицею зовнішніх стандартів. Коли значення k A значно відрізняються, то за допомогою нормальної калібрувальної кривої вводиться пропорційна визначальна похибка.
Внутрішні стандарти
Щоб використовувати зовнішню стандартизацію або метод стандартних доповнень, ми повинні вміти однаково ставитися до всіх зразків і стандартів. Коли це неможливо, точність та точність нашої стандартизації можуть постраждати. Наприклад, якщо наш аналіт знаходиться в летючому розчиннику, то його концентрація збільшиться, якщо ми втратимо розчинник до випаровування. Припустимо, у нас є зразок і стандарт з однаковими концентраціями аналіту і ідентичними сигналами. Якщо обидва відчувають однакові пропорційні втрати розчинника, то їх відповідні концентрації аналіту та сигналів залишаються ідентичними. Фактично, ми можемо ігнорувати випаровування, якщо зразки та стандарти відчувають еквівалентну втрату розчинника. Однак якщо ідентичний стандарт і зразок втрачають різну кількість розчинника, то їх відповідні концентрації та сигнали вже не рівні. У цьому випадку проста зовнішня стандартизація або стандартне додавання неможливе.
Ми все ще можемо завершити стандартизацію, якщо посилаємо сигнал аналіта на сигнал іншого виду, який ми додаємо до всіх зразків та стандартів. Вид, який ми називаємо внутрішнім стандартом, повинен відрізнятися від аналіта.
Оскільки аналіт і внутрішній стандарт отримують однакове лікування, на співвідношення їх сигналів не впливає відсутність відтворюваності в процедурі. Якщо розчин містить аналіт концентрації C A і внутрішній стандарт концентрації C IS, то сигнали, обумовлені аналітом, S A, і внутрішнім стандартом, S ІС, є
\[S_A = k_A C_A \nonumber \]
\[S_{IS} = k_{SI} C_{IS} \nonumber \]
де\(k_A\) і\(k_{IS}\) є чутливості для аналіту і внутрішнього стандарту відповідно. Прийняття співвідношення двох сигналів дає фундаментальне рівняння для внутрішньої стандартизації.
\[\frac {S_A} {S_{IS}} = \frac {k_A C_A} {k_{IS} C_{IS}} = K \times \frac {C_A} {C_{IS}} \label{sa_sis} \]
Оскільки K є співвідношенням чутливості аналіта та чутливості внутрішнього стандарту, не потрібно самостійно визначати значення для k A або k IS.
Одномісний внутрішній стандарт
При одноточковій внутрішній стандартизації ми готуємо єдиний стандарт, який містить аналіт і внутрішній стандарт, і використовуємо його для визначення значення K в Equation\ ref {sa_sis}.
\[K = \left( \frac {C_{IS}} {C_A} \right)_{std} \times \left( \frac {S_A} {S_{IS}} \right)_{std} \label{K} \]
Стандартизувавши метод, концентрація аналіта задається шляхом
\[C_A = \frac {C_{IS}} {K} \times \left( \frac {S_A} {S_{IS}} \right)_{samp} \nonumber \]
Шостий спектрофотометричний метод кількісного аналізу Pb 2+ в крові використовує Cu 2 + як внутрішній стандарт. Стандарт, який становить 1,75 ppb Pb 2 + і 2,25 ppb Cu 2 + дає співвідношення (S A/S IS) std 2,37. Зразок крові з шипами з однаковою концентрацією Cu 2 + дає коефіцієнт сигналу, (S A/S IS) samp, 1.80. Яка концентрація Pb 2 + в зразку крові?
Рішення
Рівняння\ ref {K} дозволяє обчислити значення K, використовуючи дані для стандарту
\[K = \left( \frac {C_{IS}} {C_A} \right)_{std} \times \left( \frac {S_A} {S_{IS}} \right)_{std} = \frac {2.25 \text{ ppb } \ce{Cu^{2+}}} {1.75 \text{ ppb } \ce{Pb^{2+}}} \times 2.37 = 3.05 \frac {\text{ppb } \ce{Cu^{2+}}} {\text{ppb } \ce{Pb^{2+}}} \nonumber \]
Концентрація Pb 2 +, отже, становить
\[C_A = \frac {C_{IS}} {K} \times \left( \frac {S_A} {S_{IS}} \right)_{samp} = \frac {2.25 \text{ ppb } \ce{Cu^{2+}}} {3.05 \frac {\text{ppb } \ce{Cu^{2+}}} {\text{ppb } \ce{Pb^{2+}}}} \times 1.80 = 1.33 \text{ ppb } \ce{Pb^{2+}} \nonumber \]
Кілька внутрішніх стандартів
Одноточкова внутрішня стандартизація має ті ж обмеження, що і одноточкова нормальна калібрування. Для побудови внутрішньої стандартної калібрувальної кривої ми готуємо ряд стандартів, кожен з яких містить однакову концентрацію внутрішнього стандарту та різні концентрації аналіту. У цих умовах калібрувальна крива (S A/S IS) std проти C A є лінійною з нахилом K/ C IS .
Хоча звичайна практика полягає в підготовці стандартів так, щоб кожен містив ідентичну кількість внутрішнього стандарту, це не є вимогою.
Сьомий спектрофотометричний метод кількісного аналізу Pb 2+ в крові дає лінійну внутрішню норму калібрувальної кривої, для якої
\[\left( \frac {S_A} {S_{IS}} \right)_{std} = (2.11 \text{ ppb}^{-1} \times C_A) - 0.006 \nonumber \]
Що таке ppb Pb 2 + в зразку крові, якщо (S A/S IS) samp дорівнює 2,80?
Рішення
Для визначення концентрації Pb 2 + в зразку крові замінюємо (S A/S IS) std в калібрувальному рівнянні на (S A/ S IS) samp і вирішити для C A.
\[C_A = \frac {\left( \frac {S_A} {S_{IS}} \right)_{samp} + 0.006} {2.11 \text{ ppb}^{-1}} = \frac {2.80 + 0.006} {2.11 \text{ ppb}^{-1}} = 1.33 \text{ ppb } \ce{Pb^{2+}} \nonumber \]
Концентрація Pb 2 + в зразку крові становить 1,33 ppb.
За деяких обставин неможливо підготувати стандарти так, щоб кожен містив однакову концентрацію внутрішнього стандарту. Це той випадок, наприклад, коли ми готуємо зразки по масі замість обсягу. Ми все ще можемо підготувати калібрувальну криву, однак, шляхом побудови графіка\((S_A / S_{IS})_{std}\) проти C A/C IS, даючи лінійну калібрувальну криву з нахилом K.
Ви можете задатися питанням, чи можна включити внутрішній стандарт у метод стандартних доповнень для виправлення як матричних ефектів, так і для неконтрольованих варіацій між зразками; ну, відповідь так, як описано в статті «Стандартний аналіз розведення», повне посилання на який є Джонс, В.Б.; Донаті, Г.Л.; Калловей, К.П.; Джонс, Б.Т. анал. Хім. 2015, 87, 2321-2327.