2.4: Хронічні нейродегенеративні захворювання
- Page ID
- 72928

Альфа-синуклеїнопатії та хвороба Паркінсона
Сучасна модель для більшості нейродегенеративних станів полягає в тому, що нейрони гинуть через апоптоз або запрограмовану форму загибелі клітин. Це має дуже важливі клінічні наслідки, оскільки включення або вимкнення конкретних генів/білків є методом зупинки або, можливо, зворотної нейродегенерації. Нейродегенеративні захворювання, такі як хвороба Паркінсона та хвороба Альцгеймера та інші, як вважають, використовують апоптотичні шляхи і додатково припускають, що є компонент білка, який аберрантно згортається для вироблення апоптозу.
Хвороба Паркінсона: огляд
Більшість випадків цього розладу виявляються спорадичними (тобто негенетичними за походженням). Дуже мало випадків, здається, мають генетичне походження (наприклад, гени, найчастіше пов'язані з генами DJ-1, Parkin (Ubiquitin E3 ligase) та альфа-синуклеїном). Хвороба Паркінсона найчастіше пов'язана з втратою «пігментованих» ядер в мозку і, як правило, передбачає втрату групи нейронів, знайдених в Substantia Nigra (рис\(\PageIndex{1}\).). Нейрони nigra substantia є дофамінергічними і пігментовані, оскільки вони містять білок меланін.
Типовим початком хвороби Паркінсона є середні та пізні стадії життя (тобто 50 і більше), хоча невеликий відсоток осіб, які знали генетичні мутації в Паркіні або альфа-синуклеїні, розвивають симптоми раніше.
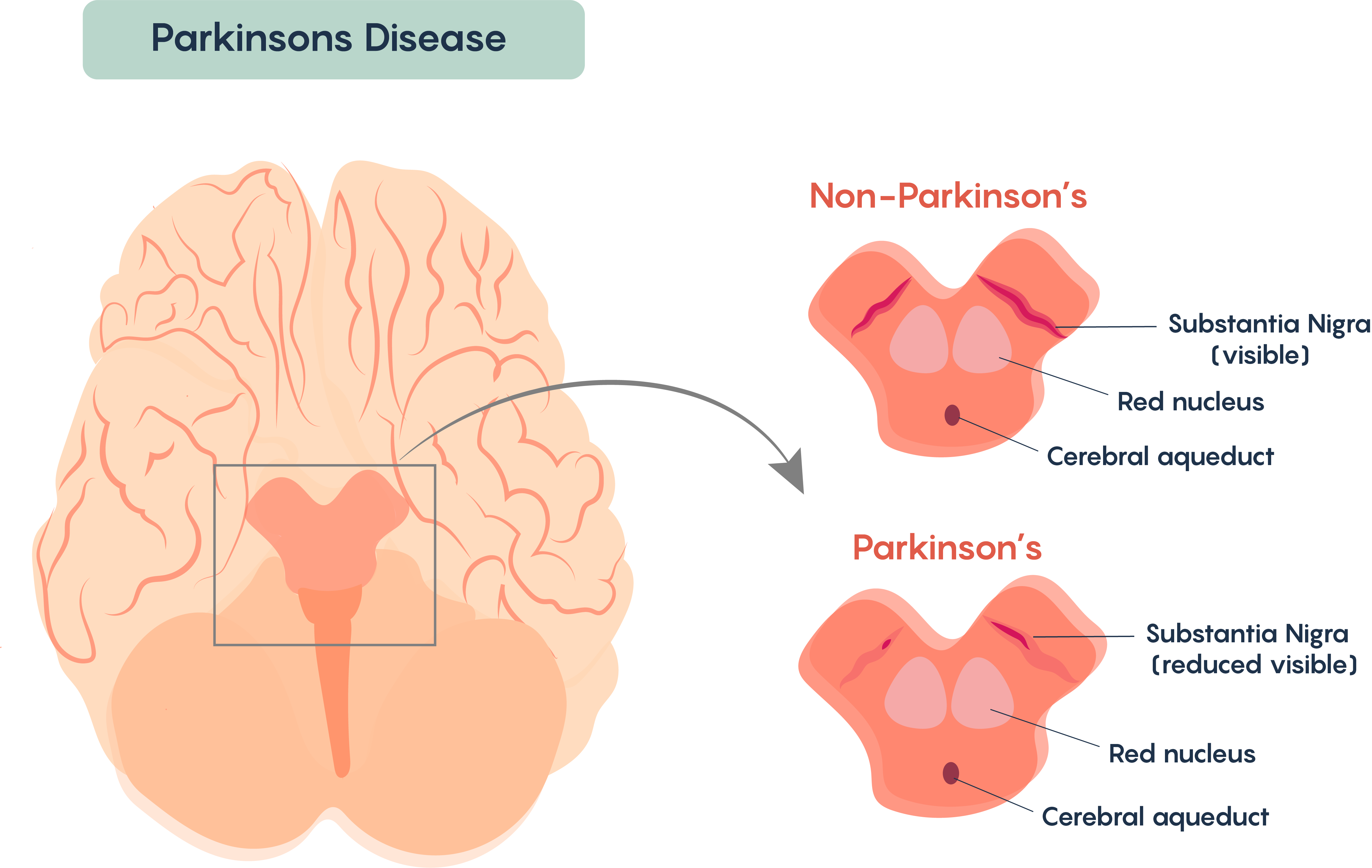
Малюнок\(\PageIndex{1}\). Втрата пігментованих дофамінергічних нейронів у межах Substantia Nigra осіб із хворобою Паркінсона.
Втрата цих клітин впливає на обробку та виконання добровільного руху у осіб з хворобою Паркінсона. Подібно до розсіяного склерозу, після діагнозу симптоми стають безперервними та прогресуючими - тобто симптоми погіршуються з часом. Знову ж таки, існує багато подібностей з розсіяним склерозом, і немає відомих ліків від хвороби Паркінсона, і хвороба залишається ідіопатичною, хоча є деякі відомі причини хвороби Паркінсона, включаючи втрату руху після церебрального атеросклерозу, вірусного енцефаліту, і в результаті боку ефекти від таких препаратів, як фенотіазиди і резерпін.
«Класичні» симптоми, пов'язані з хворобою Паркінсона
- Брадикінезія: повільність ініціації та виконання добровільних рухів
- Жорсткість: Збільшення м'язового тонусу та збільшення опору руху (руки та ноги жорсткі) - оскільки тяжкість збільшується, виробляють жорсткість зубчастого колеса
- Тремор: Зазвичай тремор у спокої; Коли людина сидить, рука трясе; Тремор зупиняється, коли людина намагається схопити щось (тремор кочення таблеток)
- Постуральна нестабільність: ненормальна фіксація постави (сутулість при стоянні), проблеми з рівновагою та рефлекс випрямлення
- Порушення ходи: Перетасування ніг
- Ортостатична гіпотензія
- Деменція (в деяких випадках)
- Дистонія (недоречне і безперервне скорочення м'язів)
- Офтальмоплегія (слабкість в очних м'язах)
- Афективні розлади настрою (такі як велика депресія)
Тіла Леві та альфа-синуклеїн — відмінні риси хвороби Паркінсона
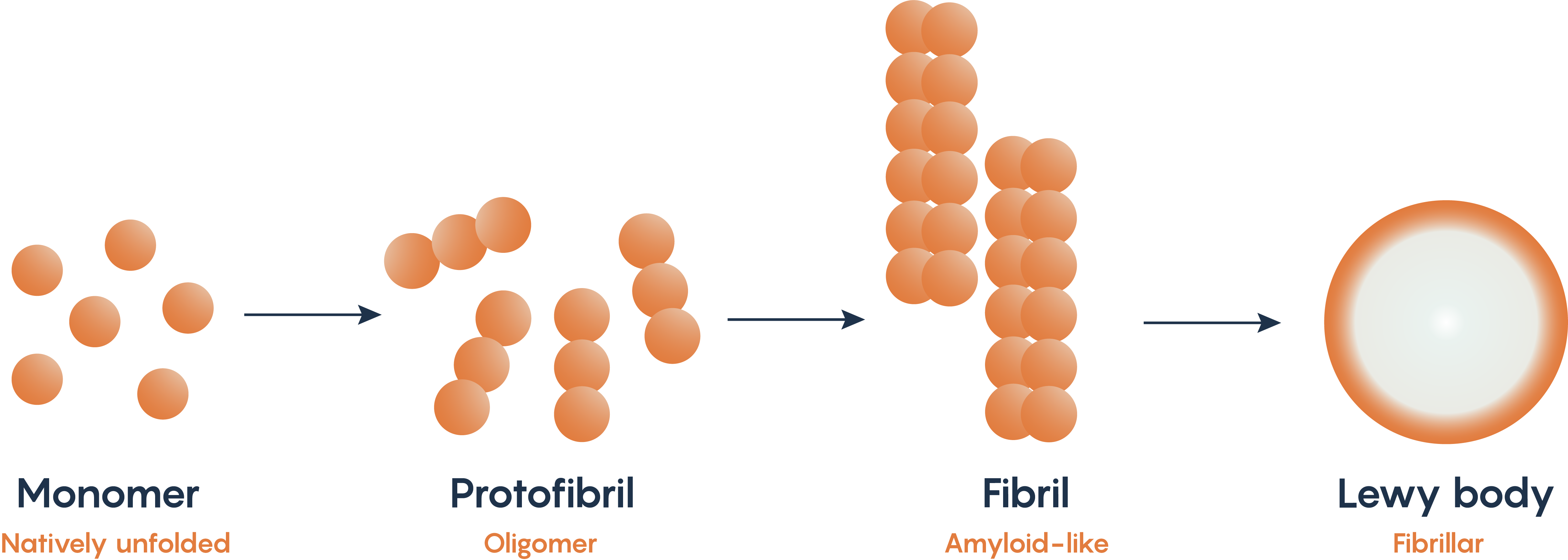
Малюнок\(\PageIndex{2}\). Неправильне згортання і подальше накопичення альфа-синуклеїну.
Альфа-синуклеїн - це природний білок всередині нейронів. Мутації в генах PARK1 і PARK4, які зазвичай кодують альфа-синуклеїн, були пов'язані з хворобою Паркінсона. Таким чином, багато моделей тварин хвороби Паркінсона шукають вироблення фібрилярної форми альфа-синуклеїну, коли він розкладається, а потім накопичується всередині субстанцій нігральних нейронів, відомих як тіла Леві (рис\(\PageIndex{2}\).). Випущено, що неправильне складання та накопичення альфа-синуклеїну є причиною того, що нейрони піддаються апоптозу, хоча точний механізм того, як це відбувається, ще належить з'ясувати.
Тваринні моделі хвороби Паркінсона
Відсутність генів-кандидатів (крім альфа-синуклеїну, Паркіна і DJ-1) означало, що більшість вчених розглянули моделі токсинів. Більшість токсинів, які виробляють функції, які нагадують зміни в русі, а також Льюї-тіло, як формування використовують катехоламінергічні руйнування. Насправді більшість цих токсинів є неймовірно потужними і небезпечними інгібіторами мітохондріального комплексу, таких як резерпін, MPTP, метамфетамін, 6-ОН-допамін, ротенон і паракват). Насправді один токсин, MPTP є побічним продуктом виробництва синтетичного героїну, що свідчить про те, що може існувати синтетична речовина, яка викликає хворобу Паркінсона, а деякі епідеміологічні дослідження показують кореляцію з використанням пестицидів (таких як паракват і ротенон) і хвороба Паркінсона.
Хвороба Альцгеймера: огляд і загальна патологія
Іншим нейродегенеративним станом є хвороба Альцгеймера (AD). Цей нейродегенеративний стан є формою деменції, що характеризується найпоширенішими симптомами, пов'язаними з хворобою Альцгеймера, зокрема втратою пам'яті, проблемами зі спілкуванням та труднощами пошуку слів, проблемами з увагою, сплутаністю та просторовою дезорієнтацією, зниженням або поганим судженням, змінами настрою і особистість.
Уражені ділянки пояснюють патологію AD, яка характеризується втратою пам'яті через усадку гіпокампу та інші проблеми, які включають більш високий рівень мислення та продуктивності, які контролюються корою. Атрофія мозку зазвичай починається в медіальній скроневій частці (тобто гіпокампі), переходить до кори асоціації і тому впливає на сенсорні та рухові області. Це також впливає на Nucleus Basalis Мейнерта, який має кілька холінергічних проекцій на кору (і вважається відповідальним за контроль сну, уваги та свідомості), як зазначено на малюнку\(\PageIndex{3}\).
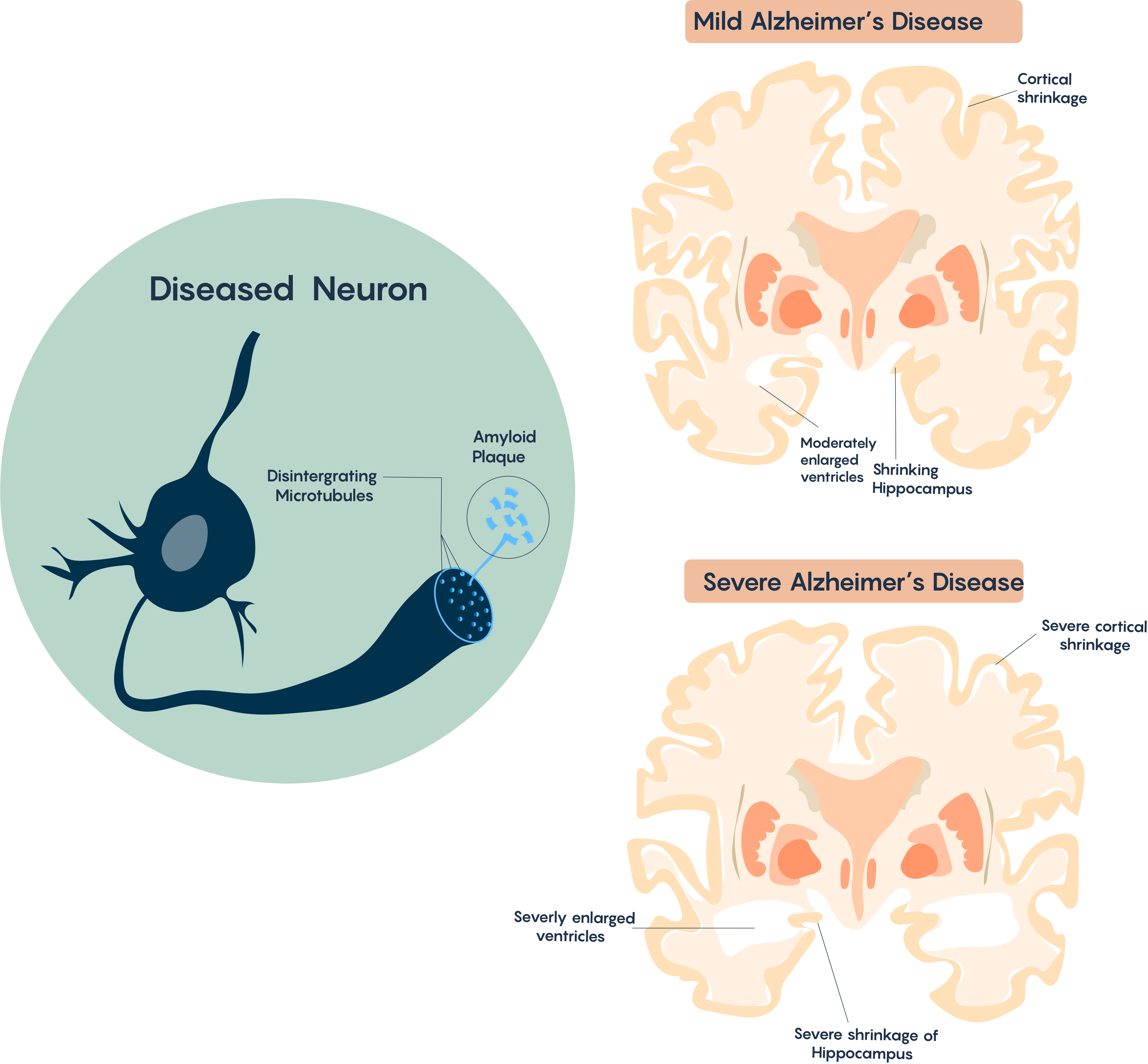
Малюнок\(\PageIndex{3}\). Усадка і випадання конкретних ділянок мозку.
Клітинна патологія
Типовий головний мозок АТ характеризується втратою нейронів, прогресуючою втратою синапсів і холінергічних проекцій, накопиченням позаклітинних β-амілоїдних (Aβ) бляшок і внутрішньоклітинних нейрофібрилярних клубів (НФТ). При збільшенні часу і патології це часто буде корелювати з утворенням гліального рубця (за участю реактивних астроцитів). Є також дуже вагомі докази того, що цей розлад також може мати сильний імунний компонент, оскільки часто відбувається інфільтрація мікрогліальних клітин у мозку хвороби Альцгеймера.
β-амілоїдні (Aβ) бляшки: βa Каскадна гіпотеза
Однією з характерних гістопатологічних і молекулярних особливостей хвороби Альцгеймера є наявність позаклітинних агрегатів, до складу яких входять -амілоїдні бляшки. Вважається, що ці бляшки індукують цитотоксичність, порушуючи нормальні функції нейронів, концентрації іонів, генерацію потенціалу дії. Як і в сучасній теорії нейродегенерації, ці бляшки складаються з розщепленої форми протеїна-попередника амілоїду (APP).
APP можна обробляти двома різними шляхами:
- Неамілоїдогенний шлях (розщеплений α- і γ-секретазами)
- Амілоїдогенний шлях (розщеплений β- і γ-секретазами)
Обробка APP
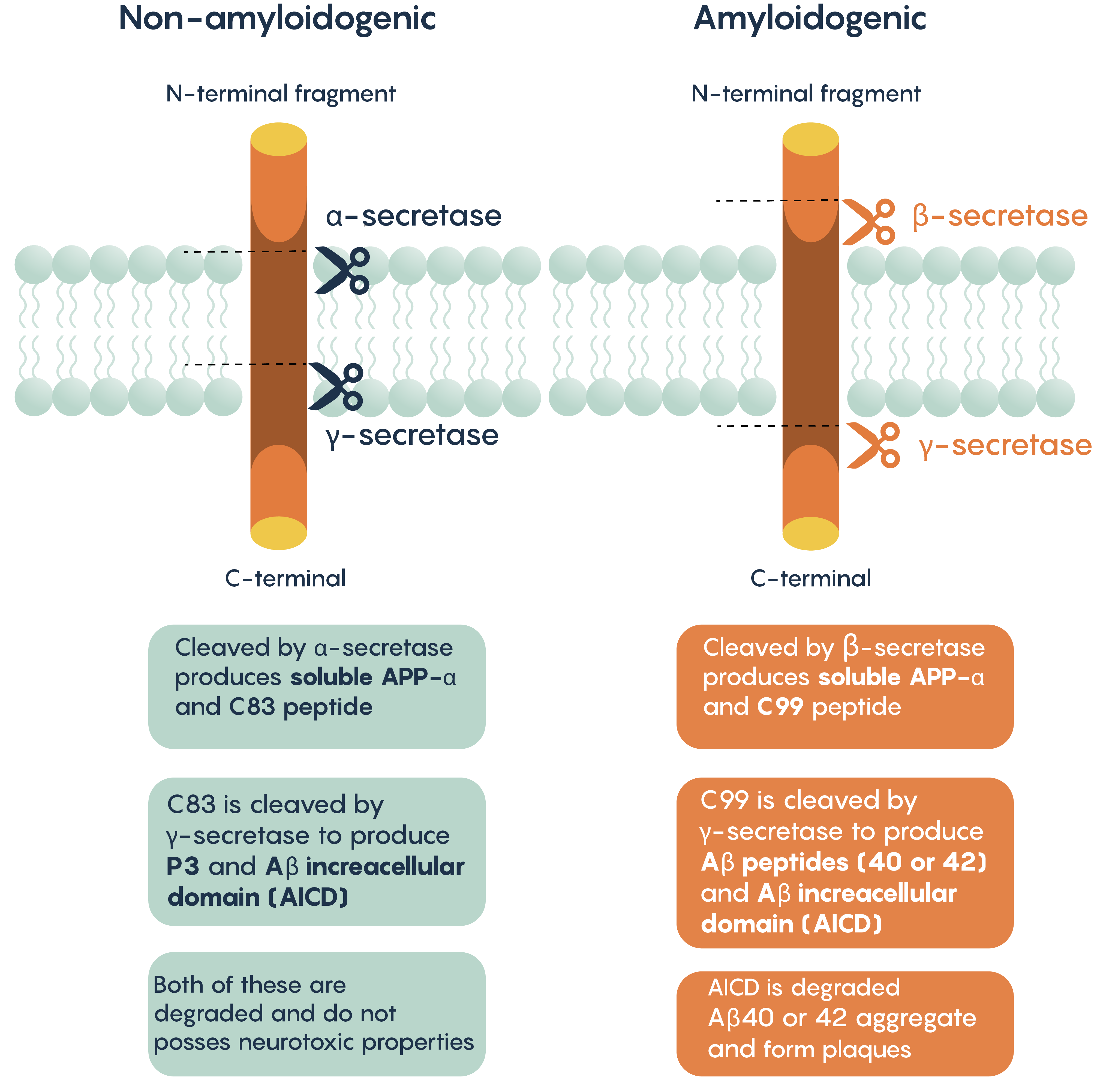
Малюнок\(\PageIndex{4}\). Шляхи обробки APP, що ведуть до виробництва та агрегації β-амілоїду.
Нейрофібрилярні клубки (NFT)
Ще однією особливістю головного мозку Альцгеймера є поява нейрофібрилярних клубів (NFT), які складаються з гіперфосфорильованого тау-білка. У нормі всередині нейронів (а насправді більшості клітин) тау зв'язується і стабілізує мікротрубочки (МТС), однак, коли це фосфорилюється кіназами, тау втрачає свою спорідненість до МТ і дисоціює з комплексу МТ. Це призводить до того, що мікротрубочки, в свою чергу, розбираються, що заважає правильному аксональному транспорту і врешті-решт призводить до втрати цілісності нейронів. Таким чином, білки фосфо-ТАУ утворюють внутрішньоклітинні агрегати (НФТ), які стають ще однією визначальною ознакою цього захворювання, а також потенційною мішенню для терапевтичного втручання.
Генетика хвороби Альцгеймера
Хоча спорадичні випадки АТ складають більшість пацієнтів з АД, сімейна АД (FAD) становить лише 5% з них. Сімейна форма, FAD проявляється раніше (близько 40-50 років) порівняно з >65 років у спорадичних випадках. Незважаючи на цю різку різницю в початку захворювання, симптоми ідентичні. Лише 50% випадків FAD можна пояснити відомими мутаціями в генах, що кодують APP і presenilins 1 і 2.
Мутації APP відносно рідкісні і характеризуються особами, які проявляють симптоми із середнім віковим початком на початку 50-х років Висновок про те, що мутації APP можуть викликати хворобу Альцгеймера, базується на спостереженні, що більшість пацієнтів з синдромом Дауна також розвивають AD після 40 років, оскільки вони мають додатковий копія Ch21 (які коди для APP). Однак більшість успадкованих форм мутацій APP змінюють обробку APP на:
- збільшити розщеплення за допомогою β-секретазного шляху
- збільшити співвідношення Aβ42/40
- тому призводять до вироблення пептидів з більш високим фібрилогенним потенціалом (рис\(\PageIndex{2}\).)