6.5: Хінін
- Page ID
- 18685

Біосинтез алкалоїдів з секологаніна
Порівняння структур поліциклічних алкалоїдів з безліччю топологічно різних скелетів, таких як преакуамміцин (317), катарантин (318) та таберсонін (319), передбачає біосинтетичну стратегію, яка збирає ці гетеромультицикли шляхом об'єднання аміноетиліндол вихідного матеріалу 400 з десятьма додатковими скелетними вуглецями. Аміноетиліндол, триптамін (400), розумно походить від декарбоксилювання амінокислоти L-триптофану (259) шляхом процесу, аналогічного декарбоксилювання тирозину (7), розглянутого в розділі 6.4.
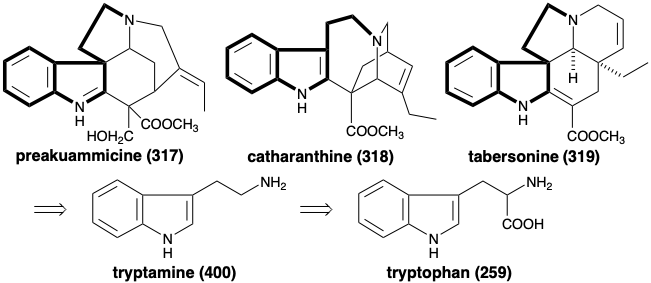
Походження решти десяти скелетних вуглеців менш очевидне. Чудовий факт, що ці залишки вуглецю мають спільне походження, добре ілюструється дослідженнями еволюції часу виробництва алкалоїдів у проростаючих проростках Vinca Rosea. 16 Таким чином, алкалоїди 317 - 326 і 328 всі ізольовані від цієї рослини, тоді як 327 є передбачуваним загальним проміжним продуктом для генерації 318 і 319 двома різними 2\(\pi\) + 4\(\pi\) циклодоповненнями.
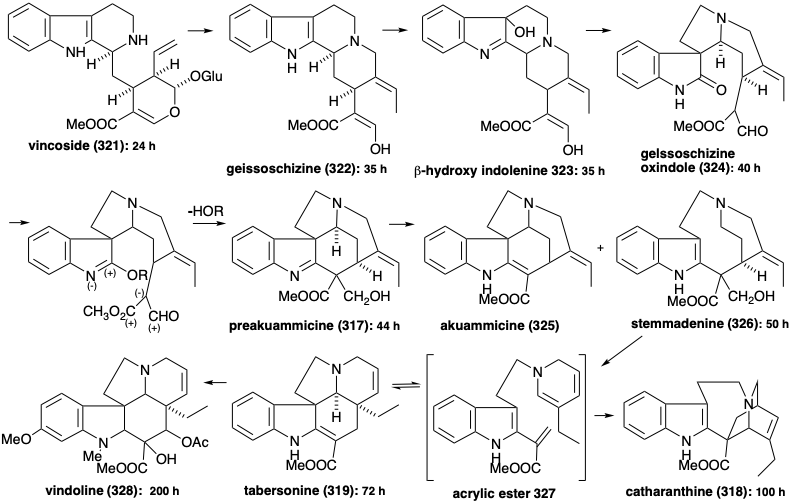
У ранньому проміжному вінкозиді (321) існує приголосний контур, який з'єднує два азоти. Два полярних відключення в цьому контурі виявляють попередник альдегіду, який мало схожий на монотерпен, крім його десяти скелетних атомів вуглецю. Проте, цим альдегідом є секологанін, терпеноїдне походження якого обговорювалося в главі 4 (див. Розділ 4.4).
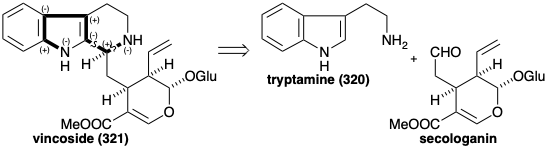
Біосинтез понад 1,000 індолних алкалоїдів з триптофану (259) починається з реакції Манніха між триптаміном (320) та секологаніном для отримання вінкозиду (321). Гідроліз глюкозиду 321 і деталізацію одержуваного гемікеталу 329 дозволяє аміно-альдегід 330. Циклізація останнього дає похідне імінію 331. Внутрішньомолекулярне додавання Michael нуклеофіла кисню з подальшим зниженням дає інший ізольований продукт, амаліцин (332). Як варіант, скорочення 331 дозволяє 322.
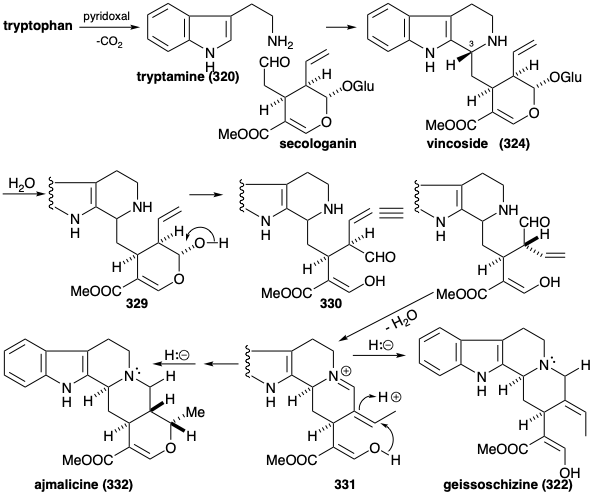
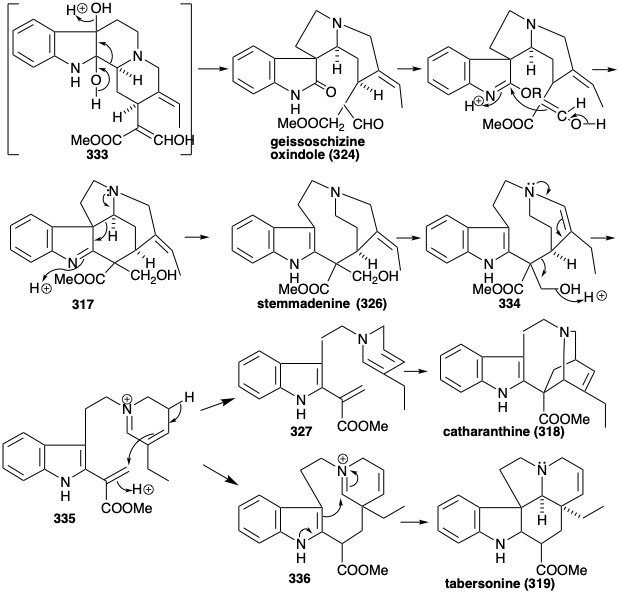
Генерація інших скелетних типів з 322 передбачає перестановки, які забезпечуються окислювальним введенням віцинального діолового масиву для отримання 333. Цей віцинальний гідроксилювання здійснюється за допомогою поетапного процесу через гідратацію ізольованого проміжного продукту, β-гідроксиіндоленіндоленіну 323. А перестановка пінаколу 333 виробляє 324. Перетворення в ефір іміно наділить 324 з реакційною здатністю, необхідною для формування преакуамміцину (317) шляхом циклізації та зменшення. Ретро-алдол-подібна фрагментація 317 з подальшим зменшенням отриманого імміну дає стеммаденін (326). Друга фрагментація ізомеру енаміну 334 з 326, мабуть, виробляє проміжний проміжний ефір акрилового ефіру 335. Діенамін таутомер 327 з 335 забезпечує ібога алкалоїдний скелет катарантину (318) внутрішньомолекулярної реакції Дільса-Альдера (не обов'язково узгодженої). В якості альтернативи, аспідосперма алкалоїдний скелет таберсонін (319) виникає з акрилового ефіру 335 за допомогою поліенової циклізації до 336 і подальшої алдол-подібної циклізації останнього з подальшою втратою протона, щоб дозволити собі таберсонін (319).
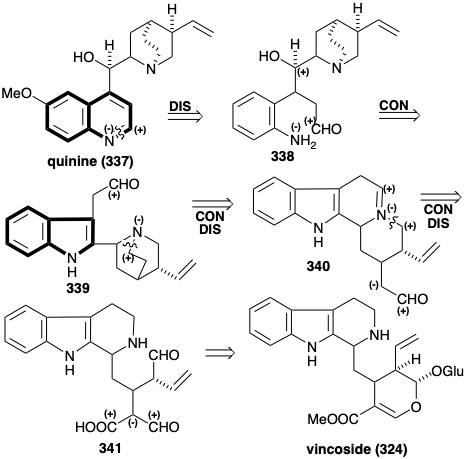
Біосинтез хініну
Ще більш загадковим є біосинтез декількох алкалоїдів, що містять гетероциклічну кільцеву систему хіноліну, таку як хінін (337). Дивовижний факт, що вуглецеві скелети цих алкалоїдів також походять з триптофану та секологаніну, додатково ілюструє довжину, до якої Природа повинна йти для досягнення біосинтезу деяких природних продуктів завдяки обмеженому інвентаризації доступних вихідних матеріалів. Це повчальна вправа зробити висновок про біосинтетичний шлях шляхом ретросинтетичного аналізу. Враховуючи граничну умову попередника індолу, має бути сформовано піридинове кільце хінолінової кільцевої системи в 337 році. Це може бути досягнуто шляхом дегідрування іміну, отриманого з попередника аміноальдегіду 338. Функціональність алкоголю в 338 може бути залишком електрофільної активуючої функціональності в попереднику, який брав участь у зв'язку з нуклеофільної аміногрупою в піррольному кільці, як у триптофану 339. Посилаючись на граничну умову триптофану як біосинтетичного вихідного матеріалу, альдегід у попереднику 339 може генеруватися гідролітичним розщепленням похідного іміну 340 бічного ланцюжка етиламіну триптаміну. Супутнє відключення одного зв'язку з третинної аміногрупою в 339 потрібно, щоб звільнити місце для підключення. Посилаючись на граничну умову вінкозиду як вихідного матеріалу, 340 може виникнути з діальдегіду 341 акарбоксилу шляхом полярного декарбоксилювання та гетероциклізації.
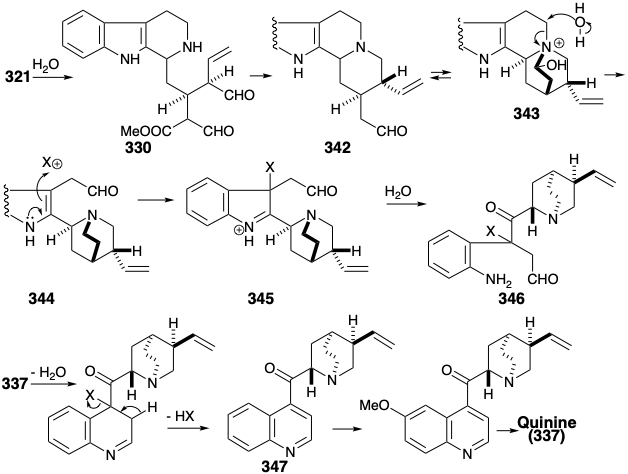
Біосинтез хінуклідинової частини хініну (337) з секологанінової частини вінкозиду (321) включає гідроліз глюкозиду з отриманням діальдегіду 330, з подальшим внутрішньомолекулярним відновним алкілуванням та декарбометиоксилюванням, щоб дати 342 , Тобто в рівновагу з геміамінальним 343. Гідролітична фрагментація останнього супроводжується окисленням первинного спирту до альдегіду і відновленням геміаміну до аміну дозволяє 344. Перестановка індолної частини 344 до хінолінового скелета ініціюється окисленням до 345 з подальшим гідролізом кільцевого розщеплення та рециркуляцією отриманого аміноальдегіду 346. Окислення арени, метилювання, а потім відновлення отриманого похідного хіноліну 347 забезпечує хінін (337).
Релейний синтез хініну
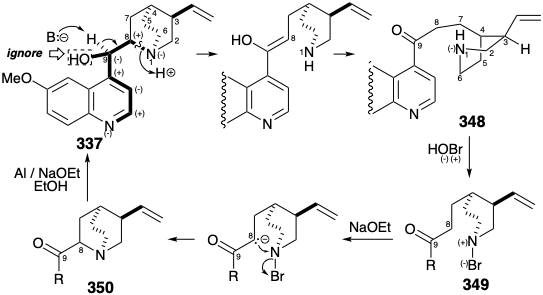
Велике топологічне спрощення хінінного скелета виникає через відключення зв'язку між атомами 1 і 8. Атом 1 є загальним атомом мультициклічної частини хінуклідину 337. Хоча атом 8 є непоширеним атомом, його роль як сполучна ланка між двома основними частинами 337 рекомендує видалити скелетні зв'язки з цим атомом. Це відключення фактично було досягнуто Rabe під час деградаційних досліджень структури 337. 17 Фрагментація від 337 до 348 залежить від полярної активації, що забезпечується аміногрупами хіноліну та хінуклідину (ігноруючи активацію, що забезпечується гідроксилом С-8).
Полярні окислювально-відновні реакції
Рабе також продемонстрував, що зворотний процес, синтез 331 з 347, може бути досягнутий шляхом використання полярної активації С-8 в 347 (нумерація хініну). 17 Цей підхід вимагає електрофілу азоту і передбачає окислення через полярні проміжні продукти. Таким чином, нуклеофільна вторинна аміногрупа в 348 перетворюється в електрофіл в 349 шляхом додавання більш електронегативного атома, тобто брому. Це являє собою окислення аміногрупи. Електрофільна атака на вуглець в 349 щоб дати 350 виробляє зв'язок між вуглецем і більш електронегативним атомом (тобто азотом). Це являє собою окислення вуглецю в поєднанні зі зменшенням аміногрупи. Ми будемо називати такі реакції, як полярні окислювально-відновні реакції. Приклад цього типу реакції зустрічається в біосинтезі ацетил CoA (див. Розділ 2.3) під час нуклеофільної атаки гідроксиетиліденом ТЕС (351) на дисульфід 352. Таким чином, нуклеофільний вуглець окислюється від f = 1 в 351 до f = 2 в 353, тоді як атом сірки в 352 відновлюється з f = -1 до f = -2 в 353. Ця реакція є складною, оскільки інший вуглець у 351 одночасно окислюється від f = 2 до f = 3 в 353 у поєднанні з відновленням другої сірки в 352 від f = -1 до f = -2 в 353.
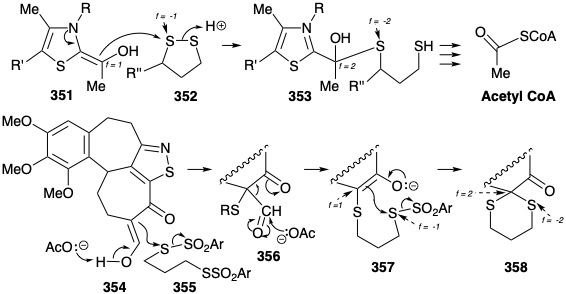
Інша полярна окислювально-відновна реакція, знову ж таки з сіркою як електрофіл, зустрічалася в синтезі Вудворда колхіцину (див. Розділ 6.1). Фактично введення дитіокеталу в нуклеофільний вуглець α до карбонільної групи включає два послідовних окислення α вуглецю, спочатку від 354 до 356, потім останнього в 358, в поєднанні з двома відновленнями сірки, спочатку в перетворенні 355 до 356 потім в перетворенні останнього, через 357, в 358. Ця реакція також є складною, оскільки інший вуглець у 354, α до енольного гідроксилу, одночасно окислюється від f = 1 до f = 2 в 356 у поєднанні з відновленням другої сірки в 355, а другий вуглець окислюється з f = -1 в 357 до f = -2 в 358 (карбонільний вуглець) в поєднанні з відновленням другої сірки.
Конвергентна стратегія для ключового проміжного класу 348
Слід зазначити, що 350 (див. Вище) являє собою суміш епімерів при С-8, а скорочення, яке виробляє 337, вводить ще один асиметричний центр (при С-9). На щастя, 337 був основним компонентом ізомерної суміші, отриманої цим нестереоконтрольованим перетворенням 348 до 337. Ця конверсія робить хінотоксин (348) привабливою субмішенню для загального синтезу хініну (337). Субмішень 348 додатково спрощується дислокацією, яка розбиває молекулу на два великі фрагменти шляхом розриву однієї з чотирьох зв'язків, що з'єднують кільця хіноліну та піперидину. Вивих, обраний Вудвордом і Дерінгом для першого загального синтезу хініну (337), був продиктований тим, що зворотний процес, синтез 348 з 359 і 360, мав відмінний прецедент. Дигідро похідне 348 (з етилом замість вінілової групи) було отримано Rabe з 359 і дигідро похідне 360, яке було отримано в результаті деградації природного хініну (337).

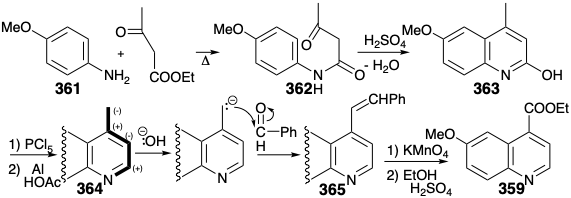
Загальні синтези субмішені етилхінінат (359) також були відомі, коли був здійснений загальний синтез 337. Особливо ефективний спосіб вводить карбоксильну групу у прихованій формі як бензилову метильну групу та конструює гетероцикл азоту на попередньо сформованому ароматичному попереднику 361 шляхом полярних реакцій. Циклодегідратація 362 дозволяє 363, тобто знижується до 364. Бензилове окислення 364 досягається окислювальним розщепленням прихованої карбонової кислоти, зв'язку C = C в попереднику 365, яка доступна шляхом полярної конденсації 364 з бензальдегідом. Конденсація використовує нуклеофільну активацію бензилового метилу, що забезпечується азотом у 364 році.
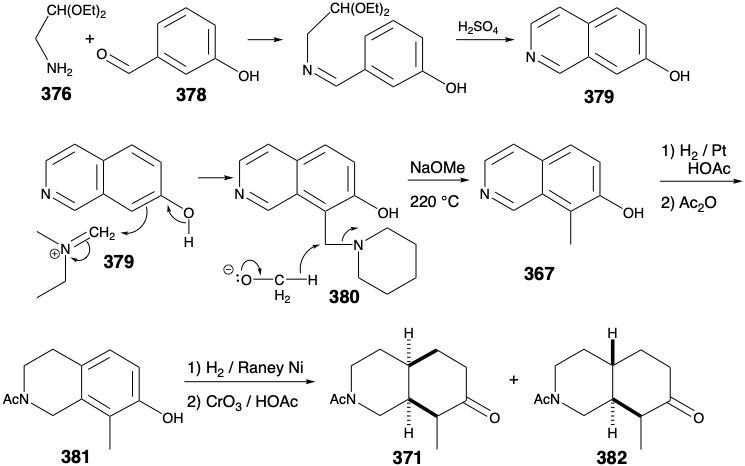
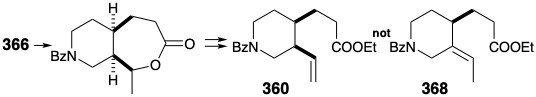
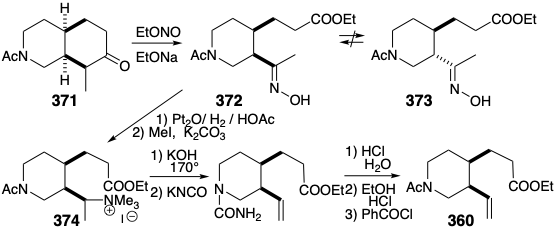
Єдиною синтетичною метою, що залишилася, таким чином, був етил N-бензоїлгомомерохінінат (360). Дві бічні ланцюги в 360 можуть бути створені стереоспецифічно цис шляхом окислювального розщеплення тимчасового моста в 366. Цис кільце злиття в 366 може бути вироблено, в свою чергу, шляхом каталітичного гідрування ароматичного попередника ізохіноліну 367.

Необхідно уникати кількох потенційних недоліків при розробці детальної схеми перетворення 366 на 360. Наприклад, розщеплення тимчасового моста в 366 шляхом окислення Байєра-Віллігера з подальшим ліквідацією для генерації вінілової групи в 360 повинна уникати утворення альтернативного, термодинамічно сприятливого, похідного етилідену 368.
Цікаво, що реакція, обрана для досягнення кільцевого розщеплення, була реакцією, яка використовувалася раніше в дослідженнях деградації для визначення структури хініну. Так, Рабе здійснив розщеплення хінінону (369) на етилхінінат (359) і оксиміносполуку 370 шляхом обробки амілнітратом і етоксидом натрію. Це розщеплення аналогічно ретро-клайсену реакції, яка особливо легко відбувається для неенолізуваних β-кето-ефірів.
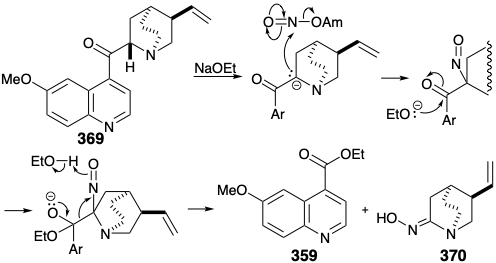
Застосування цього процесу розщеплення до N-ацетилового аналога 371 з 366 генерує оксим 372. Потенційний недолік, епімеризація цис-1,2-дизаміщеного продукту 372 в термодинамічно більш стабільний транс-ізомер 373, не була проблемою. На відміну від того, що очікувалося б для відповідного похідного ацетилу, оксим 372 не схильний до епімеризації. Таким чином, гідроксильний протон, а не α-протон переважно абстрагується при обробці оксимів основою. Зменшення та метилювання 372 легко забезпечує четвертинне похідне амонію 374, що забезпечує необхідну термінальну вінілову групу в 360 за базою сприяла ліквідації Хофмана за участю регіоселективного абстрагування водню з менш заміщеного β вуглецю.
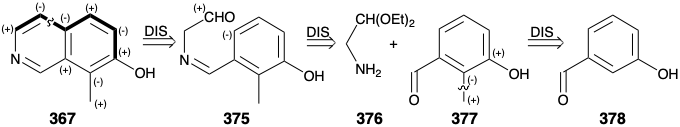
Як зазначалося вище, для синтезу етилу N- бензоїлгомомерохінінату (360) був обраний ароматичний попередник 367. Моноциклічний ароматичний попередник для 367 може бути або похідним піридину, або похідним бензолу. Вибір останнього дозволяє використовувати електрофільне ароматичне заміщення на багатому електроном попереднику для здійснення відпалу піридинового кільця. Ця відпалювання вимагає вуглецево-вуглецевих зв'язків мета і пара до гідроксильної групи. Формуванню паразв'язку електрофільним ароматичним заміщенням сприяє над мета-сильним доноруючим ефектом електрононного активуючого ефекту гідроксильної групи. Утворення цього паравуглецево-вуглецевого зв'язку на останньому етапі відпалу свідчить про фенольний попередник 375 з чотирма атомами початкового піридинового кільця, приєднаними до мета-позиції. Зв'язок між цим мета-замінником та фенольним кільцем не може бути сформований електрофільною ароматичною заміщенням, оскільки віддають перевагу орто і пара, а не метазаміщення. Однак відключення цього замінника шляхом видалення зв'язку між азотом і бензиловим вуглецем свідчить про дисонантний карбоніл-маскуваний аміноацетальдегід 376 і похідне бензальдегіду 377. Метиловий замінник у 377 році також може бути введений електрофільним ароматичним заміщенням на легкодоступний м-гідрокси-бензальдегід (378). Однак досягнення необхідного регіонального контролю в такому алкілуванні може бути важко.
По суті, був прийнятий інший порядок кроків. Введення ортометилзамінника було відкладено до завершення відпалу піридинового кільця, оскільки введення метильної групи може бути легко досягнуто регіоселективно шляхом електрофільного ароматичного заміщення на β-гідроксиізохінолін 379. Таким чином, амінометилювання піперидином і формальдегідом виробляється бензиловий амін 380, який при нагріванні зменшувався до 367 в присутності метоксиду натрію. Це незвичайне скорочення передбачає перенесення гідридів з метоксиду. Технічні труднощі виникли при гідруванні 367 на 371. Таким чином, оскільки амін отруїв каталізатор, гідрування припинилося після того, як було зменшено лише кільце, що містить азот. Амін повинен був бути заблокований як амід, перш ніж можна було досягти зменшення бензольного кільця. Бажаної цис-стереоспецифічності при скороченні 381 до 371 досягти не вдалося. На щастя, однак, це не було фатальним недоліком, оскільки необхідний ізомер може бути виділений із продукту реакції трансплавленого гідрогенізації 383. Таким чином, каталітичне гідрування не зовсім надійно для стереоселективної доставки водню на одну грань ароматичного кільця.