27.2: Інструменти для газової хроматографії
- Page ID
- 27261

У газовій хроматографії (ГК) ми впорскуємо зразок, який може бути газом або рідиною, в газоподібну рухливу фазу (часто називають газом-носієм). Рухлива фаза переносить зразок через упаковану або капілярну колону, яка розділяє компоненти зразка на основі їх здатності розділяти між рухомою фазою і стаціонарною фазою. На малюнку Template:index показаний приклад типового газового хроматографа, який складається з декількох ключових компонентів: подачі стисненого газу для рухомої фази; нагрітого інжектора, який швидко випаровує компоненти в рідкому зразку; колонка, яка поміщається всередині печі, температура якої ми може контролювати під час поділу; і детектор для спостереження за елюентом, коли він відривається від колонки. Розглянемо кожен з цих компонентів.

Мобільна фаза
Найбільш поширеними рухливими фазами для газової хроматографії є He, Ar та N 2, які мають перевагу в тому, що вони хімічно інертні як до зразка, так і до стаціонарної фази. Вибір газу-носія часто визначається потребами детектора приладу. Для упакованої колони швидкість рухомої фази зазвичай становить 25—150 мл/хв. Типова швидкість потоку для капілярної колонки становить 1-25 мл/хв.
Введення зразка
Три фактори визначають, як ми вводимо зразок на газовий хроматограф. По-перше, всі складові зразка повинні бути летючими. По-друге, аналіти повинні бути присутніми у відповідній концентрації. Нарешті, фізичний процес введення зразка не повинен погіршувати поділ. Кожна з цих потреб розглядається в цьому розділі.
Підготовка летючого зразка
Не кожен зразок можна вводити безпосередньо в газовий хроматограф. Для переміщення по колоні складові зразка повинні бути досить летючими. Розчинена речовина низької летючості, наприклад, може затримуватися колоною і продовжувати елютувати під час аналізу наступних зразків. Енергонезалежний розчинений розчин буде конденсуватися у верхній частині колони, погіршуючи продуктивність колонки.
Привабливим підходом до виділення аналітів є твердофазна мікроекстракція (SPME). В одному підході, який проілюстрований на малюнку Template:index, всередині голки шприца поміщається плавлене кремнеземие волокно. Волокно, яке покрито тонкою плівкою адсорбентного матеріалу, такого як полідиметилсилоксан, опускається в зразок шляхом пригнічення плунжера і піддається впливу зразка протягом заданого часу. Після виведення волокна в голку його переносять на газовий хроматограф для аналізу.

Два додаткових методу ізоляції летких аналітів - це продувка і пастка і відбір проб. У продувці та пастці ми бульбашковим інертним газом, таким як He або N 2, через зразок, виділяючи - або продуваючи - летючі сполуки. Ці сполуки переносяться продувним газом через пастку, яка містить абсорбуючий матеріал, такий як Tenax, де вони утримуються. Нагрівання пастки і зворотна промивка газом-носієм переносить леткі сполуки на газовий хроматограф. У відборі відбору проб поміщаємо в закритий флакон з вищерозташованим повітряним простором. Після того, як летючі аналіти мали час для вирівнювання між зразком і вищевказаним повітрям, ми використовуємо шприц для вилучення частини парової фази і впорскування її в газовий хроматограф. Крім того, ми можемо взяти зразок headspace за допомогою SPME.
Термічна десорбція є корисним методом вивільнення летких аналітів з твердих речовин. Розміщуємо частину твердого тіла в скляну трубку з нержавіючої сталі. Після продувки газом-носієм для видалення будь-якого O 2, який може бути присутнім, нагріваємо зразок. Летючі аналіти змітаються з трубки інертним газом і переносяться в ГК. Оскільки випаровування не є швидким процесом, летючі аналіти часто концентруються у верхній частині колони шляхом охолодження вхідного отвору колонки нижче кімнатної температури, процес, відомий як кріогенне фокусування. Після завершення випаровування вхідний отвір колонки швидко нагрівається, звільняючи аналіти для переміщення через колону.
Причина видалення О 2 полягає в тому, щоб не допустити реакції окислення зразка при його нагріванні.
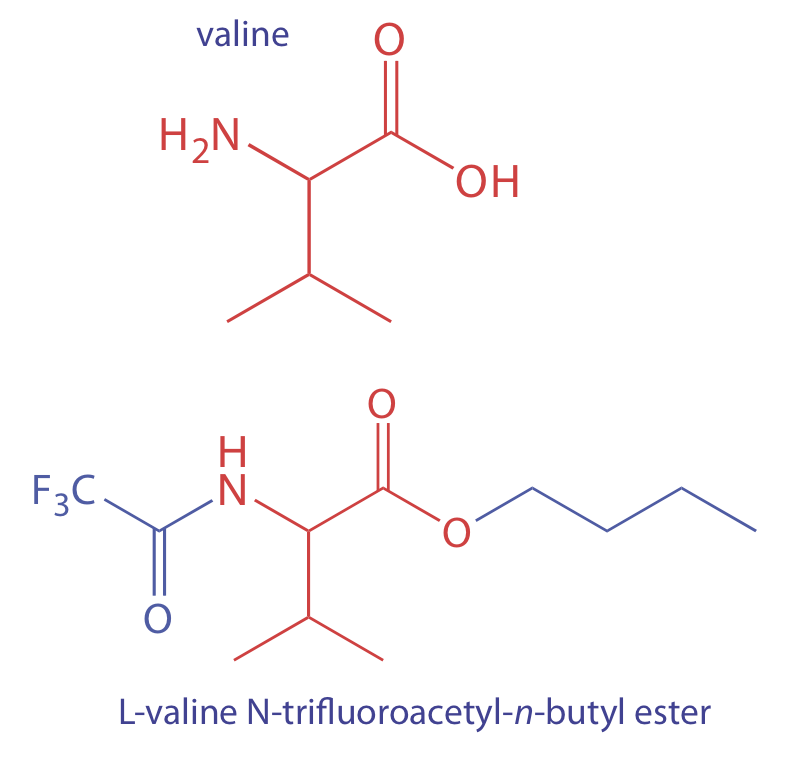
Щоб проаналізувати енергонезалежний аналіт, ми повинні перетворити його в летючу форму. Наприклад, амінокислоти недостатньо летючі для аналізу безпосередньо за допомогою газової хроматографії. Реагуючи амінокислоту, таку як валін, з 1-бутанолом і ацетилхлоридом виробляє етерифіковану амінокислоту. Подальша обробка трифтороцтової кислотою дає летке похідне N-трифторацетил-н-бутилового ефіру амінокислоти.
Регулювання концентрації аналіта
У аналіта концентрація занадто мала, щоб дати адекватний сигнал, тоді ми повинні сконцентрувати аналіт, перш ніж вводити зразок у газовий хроматограф. Побічна перевага багатьох методів видобутку полягає в тому, що вони часто концентрують аналіти. Летючі органічні матеріали, виділені з водного зразка продувкою-пасткою, наприклад, концентруються на стільки ж, скільки\(1000 \times\).
Якщо аналіт занадто концентрований, легко перевантажити колону, що призводить до пікового фронтального і поганого поділу. Крім того, концентрація аналіта може перевищувати лінійну реакцію детектора. Ін'єкція меншої кількості зразків або розведення зразка летючим розчинником, таким як метиленхлорид, є двома можливими рішеннями цієї проблеми.
Ін'єкція зразка
У розділі 26 ми розглянули кілька пояснень того, чому смуга розчиненої речовини збільшується в ширину, коли вона проходить через колону, процес, який ми назвали розширенням смуги. Ми також вводимо додаткове джерело розширення смуги, якщо не вдасться ввести зразок в мінімально можливий об'єм рухомої фази. Існує два основних джерела розширення цієї передколонної смуги: впорскування зразка в рухомий потік рухомої фази та введення рідкого зразка замість газоподібного зразка. Конструкція інжектора газового хроматографа допомагає мінімізувати ці проблеми.
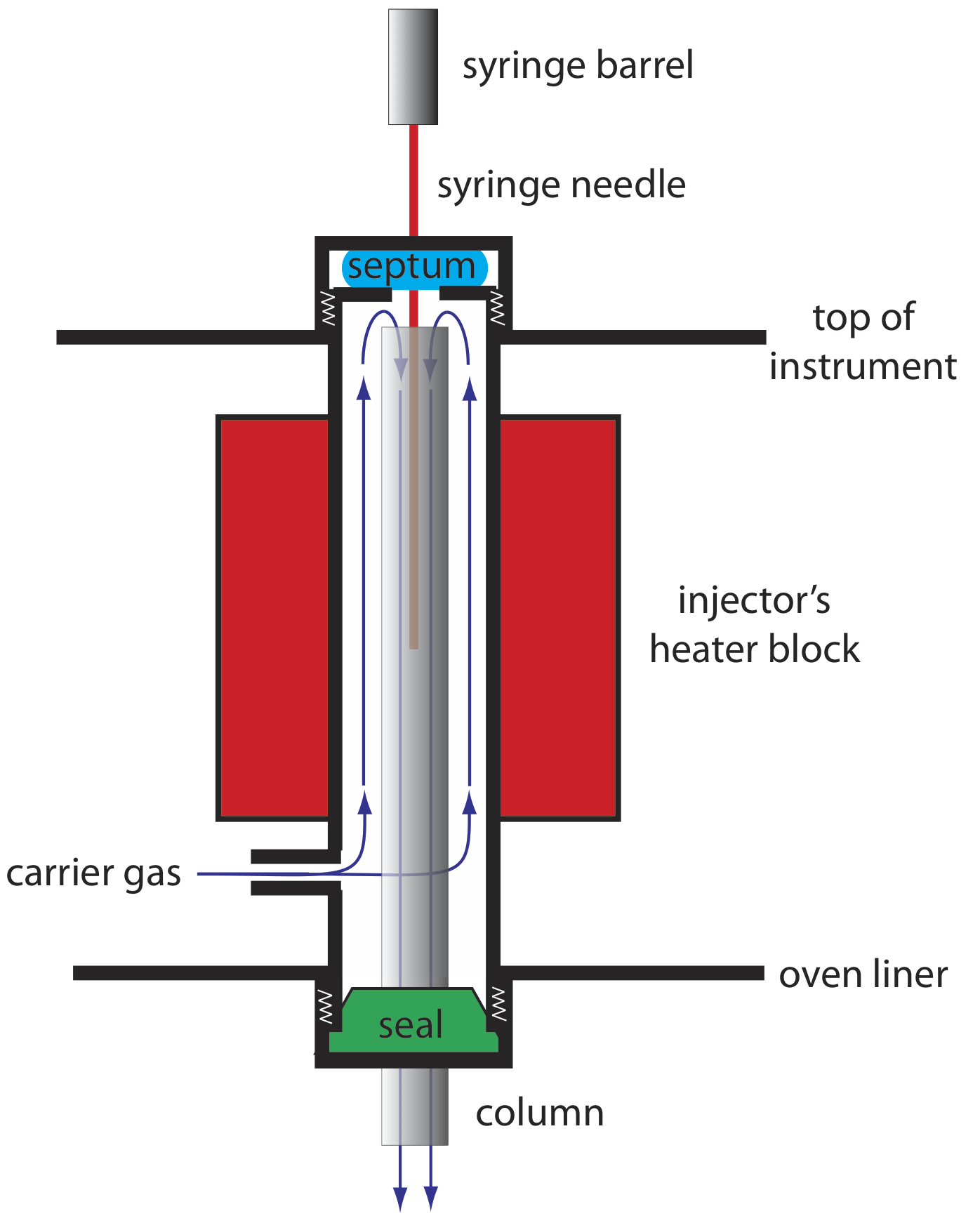
Приклад простого порту введення для упакованого стовпця наведено на малюнку Template:index. Верх колонки поміщається всередині нагрітого інжекторного блоку, з газом-носієм, що надходить знизу. Зразок вводять через гумову перегородку за допомогою мікролітрового шприца, такого як показано на малюнку Template:index. Впорскування зразка безпосередньо в колону мінімізує розширення смуги, оскільки він змішує зразок з найменшою можливою кількістю газу-носія. Блок інжектора нагрівається до температури не менше 50 о С вище температури кипіння найменш летючого розчиненого речовини, що забезпечує швидке випаровування компонентів зразка.

Оскільки обсяг капілярної колонки значно менший, ніж для упакованої колонки, він вимагає іншого стилю інжектора, щоб уникнути перевантаження колони зразком. На малюнку Template:index показано принципову схему типового розщепленного/безрозщепленого інжектора для використання з капілярною колоною.
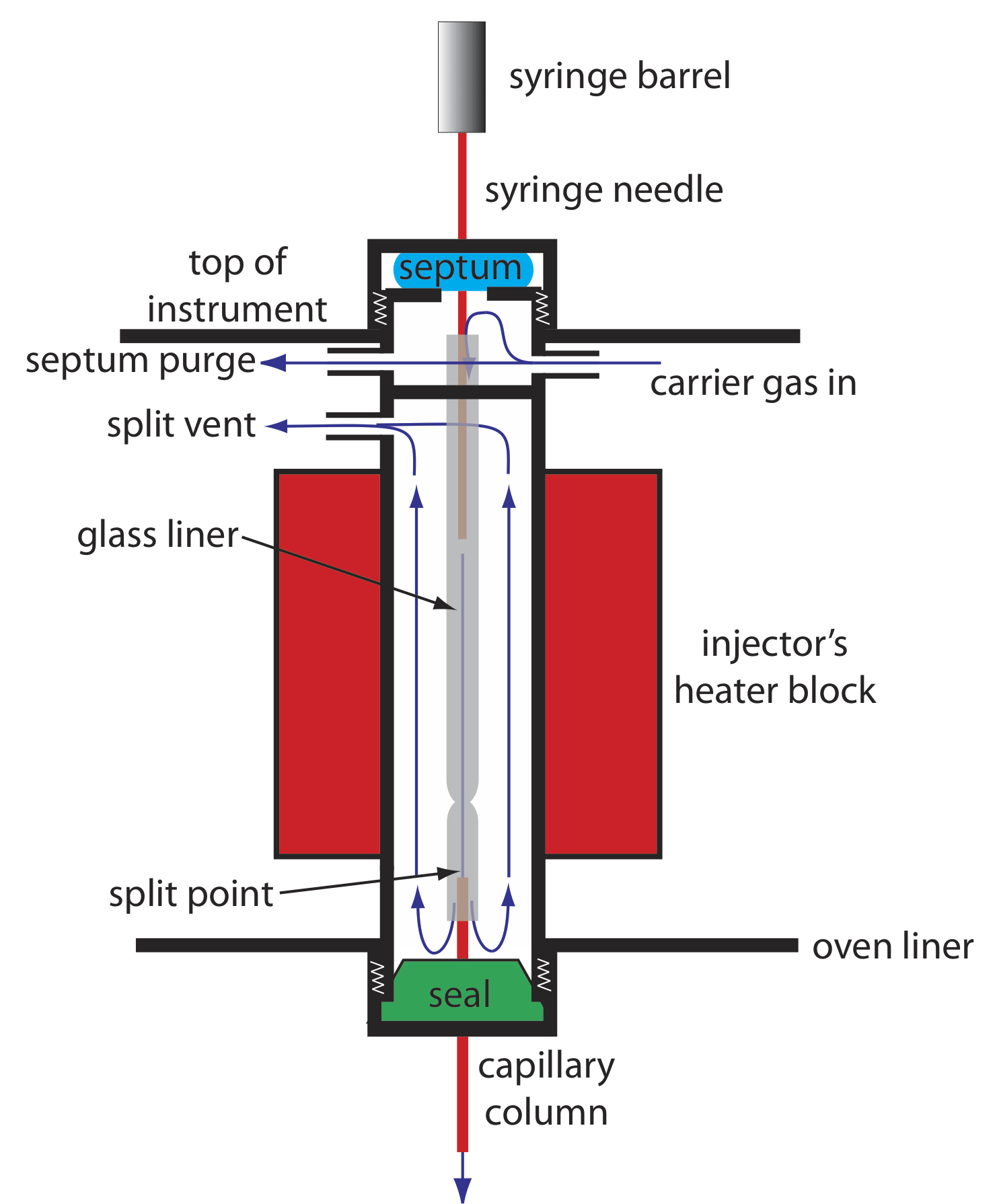
У розщепленої ін'єкції впорскуємо зразок через гумову перегородку за допомогою мікролітрового шприца. Замість того, щоб впорскувати зразок безпосередньо в колонку, він впорскується в скляний вкладиш, де змішується з газом-носієм. У точці розщеплення невелика частка газу-носія і проби надходить в капілярну колону, а залишок виходить через розщеплений вентиляційний отвір. Контролюючи швидкість потоку газу-носія, коли він надходить в інжектор, і його швидкість потоку через продувку перегородки та розділений вентиляційний отвір, ми можемо контролювати частку зразка, яка надходить у капілярну колону, як правило, 0,1— 10%.
Наприклад, якщо витрата газу-носія становить 50 мл/хв, а витрати для продувки перегородки і розщепленого вентиляційного отвору складають відповідно 2 мл/хв і 47 мл/хв, то витрата через колонку становить 1 мл/хв (= 50 - 2 - 47). Співвідношення зразка, що надходить в колонку, становить 1/50, або 2%.
У безрозрізному впорскуванні, який корисний для аналізу слідів, ми закриваємо розділений отвір і дозволяємо всьому газу-носія, який проходить через скляний вкладиш, потрапляти в колонку - це дозволяє практично всьому зразку потрапляти в колонку. Оскільки швидкість потоку через інжектор низька, значне розширення смуги преколони є проблемою. Утримання температури колонки приблизно на 20-25 o С нижче температури кипіння розчинника дозволяє розчиннику конденсуватися при вході в капілярну колону, утворюючи бар'єр, який затримує розчинені речовини. Після дозволу розчинених речовин концентруватися температуру колони підвищують і починають поділ.
Для зразків, які легко розкладаються, може знадобитися ін'єкція на колонці. При цьому способі зразок нагнітається безпосередньо в колону без нагрівання. Потім температуру стовпчика підвищують, випаровуючи зразок з такою низькою температурою, наскільки це практично.
Контроль температури
Контроль температури колонки має вирішальне значення для досягнення хорошого поділу при використанні газової хроматографії. З цієї причини колонка розміщується всередині терморегульованої печі (див. Рисунок Template:index). При ізотермічному поділі підтримуємо колону при постійній температурі. Щоб збільшити взаємодію між розчиненими речовинами та стаціонарною фазою, температуру зазвичай встановлюють трохи нижче температури самого низького кипіння розчиненого речовини.
Однією з труднощів ізотермічного поділу є те, що температура, яка сприяє відділенню низькокиплячого розчиненого речовини, може призвести до неприпустимо тривалого часу утримання для більш високого кипіння розчиненої речовини. Програмування температури забезпечує вирішення цієї проблеми. На початку аналізу ми встановили початкову температуру колонки нижче, ніж для найбільш низькокиплячого розчиненого речовини. У міру просування поділу ми повільно збільшуємо температуру або з рівномірною швидкістю, або в ряд кроків.
Детектори для газової хроматографії
Завершальною частиною газового хроматографа є детектор. Ідеальний детектор має кілька бажаних особливостей: низьку межу виявлення, лінійну реакцію в широкому діапазоні концентрацій розчинених речовин (що полегшує кількісну роботу), чутливість до всіх розчинених речовин або селективність для певного класу розчинених речовин та нечутливість до зміни швидкості потоку або температури.
Детектор теплопровідності (TCD)
Один з найбільш ранніх детекторів газової хроматографії використовує переваги теплопровідності рухомої фази. Коли рухлива фаза виходить з колони, вона проходить над вольфрамово-ренієвою ниткою дроту (див. Рисунок Template:index). Електричний опір нитки розжарювання залежить від її температури, яка, в свою чергу, залежить від теплопровідності рухомої фази. Через високу теплопровідність гелій є рухомою фазою вибору при використанні детектора теплопровідності (TCD).
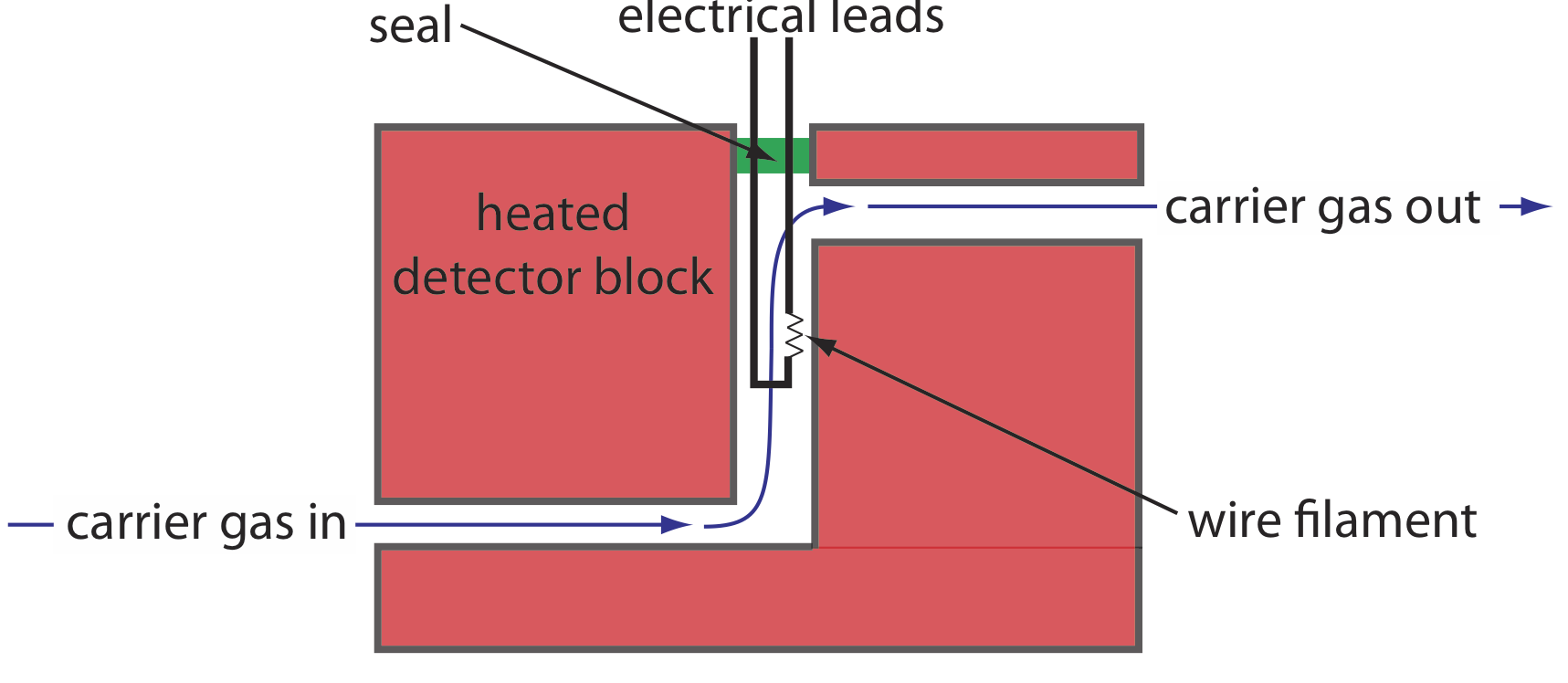
Теплопровідність, як випливає з назви, є мірою того, наскільки легко речовина проводить тепло. Газ з високою теплопровідністю відводить тепло від нитки - і, таким чином, охолоджує нитку - швидше, ніж газ з низькою теплопровідністю.
Коли розчинене речовина елюює з колони, теплопровідність рухомої фази в комірці TCD зменшується і температура нитки проводу, а значить, і її опору, збільшується. Опорна комірка, через яку проходить лише рухлива фаза, виправляє будь-які залежні від часу зміни швидкості потоку, тиску або електричної потужності, всі з яких впливають на опір нитки розжарювання.
Оскільки всі розчинені речовини впливають на теплопровідність рухомої фази, детектор теплопровідності є універсальним детектором. Ще однією перевагою є лінійна реакція TCD над діапазоном концентрацій, що охоплює 10 4 —10 5 порядків. Детектор також неруйнівний, що дозволяє відновлювати аналіти за допомогою постдетекторної холодної пастки. Одним із суттєвих недоліків детектора TCD є його погана межа виявлення для більшості аналітів.
Детектор іонізації полум'я (FID)
Горіння органічної сполуки в H 2/повітряне полум'я призводить до полум'я, яке містить електрони та органічні катіони, імовірно CHO +. Застосування потенціалу приблизно 300 вольт через полум'я створює невеликий струм приблизно від 10 до 10 -12 ампер. При посиленні цей струм забезпечує корисний аналітичний сигнал. Це основа популярного детектора іонізації полум'я, принципова схема якого представлена на малюнку Template:index.
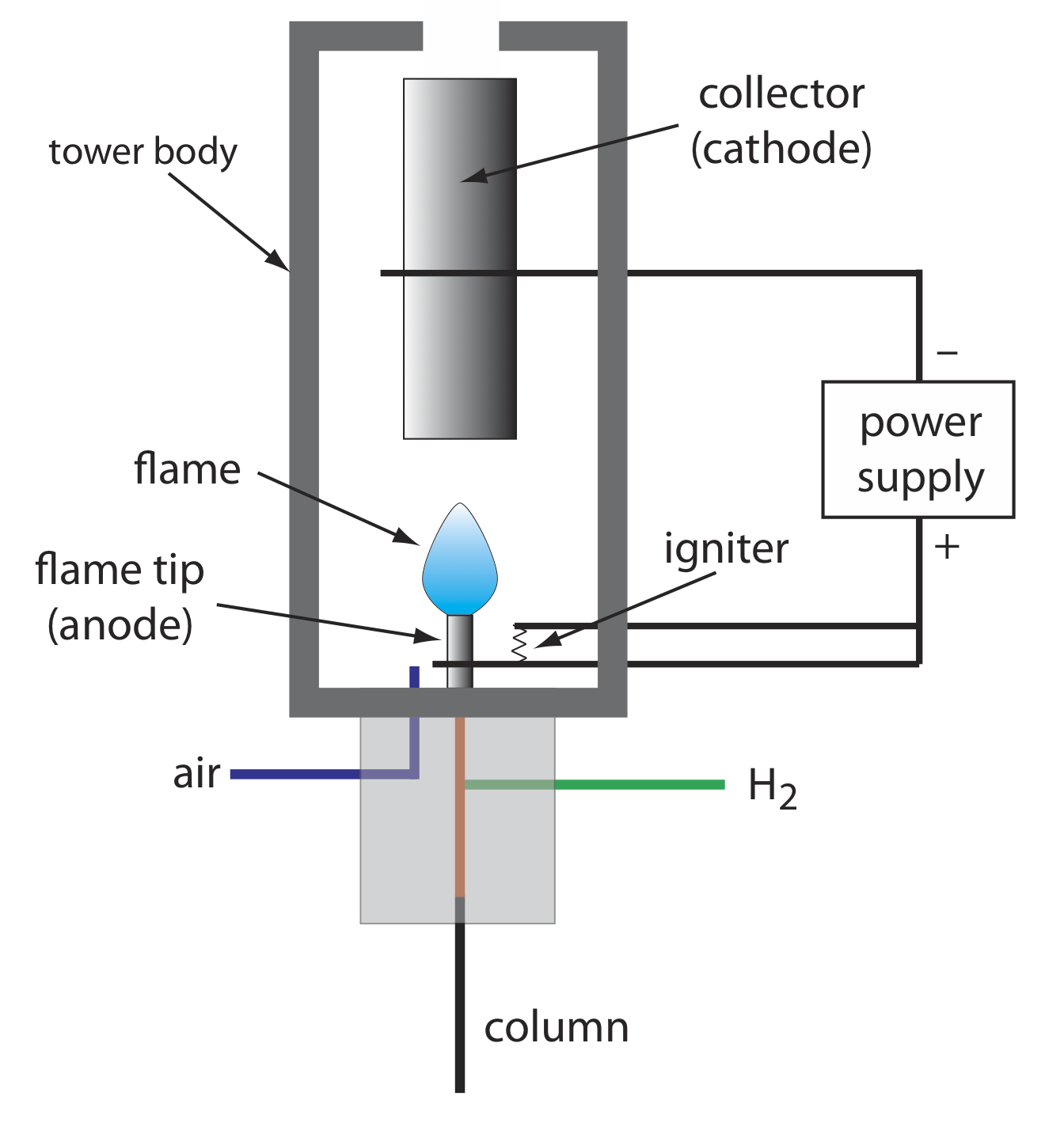
Більшість атомів вуглецю - за винятком карбонільних та карбонових груп - генерують сигнал, що робить FID майже універсальним детектором органічних сполук. Більшість неорганічних сполук і багато газів, таких як H 2 O і CO 2, не виявляються, що робить детектор FID корисним детектором для аналізу органічних аналітів в атмосферних і водних зразках навколишнього середовища. До переваг FID можна віднести межу виявлення, яка приблизно на два-три порядки менше, ніж у детектора теплопровідності, і лінійний відгук понад 10 6 —10 7 порядків у кількості введеного аналіту. Зразок, звичайно ж, руйнується при використанні датчика іонізації полум'я.
Детектор захоплення електронів (ECD)
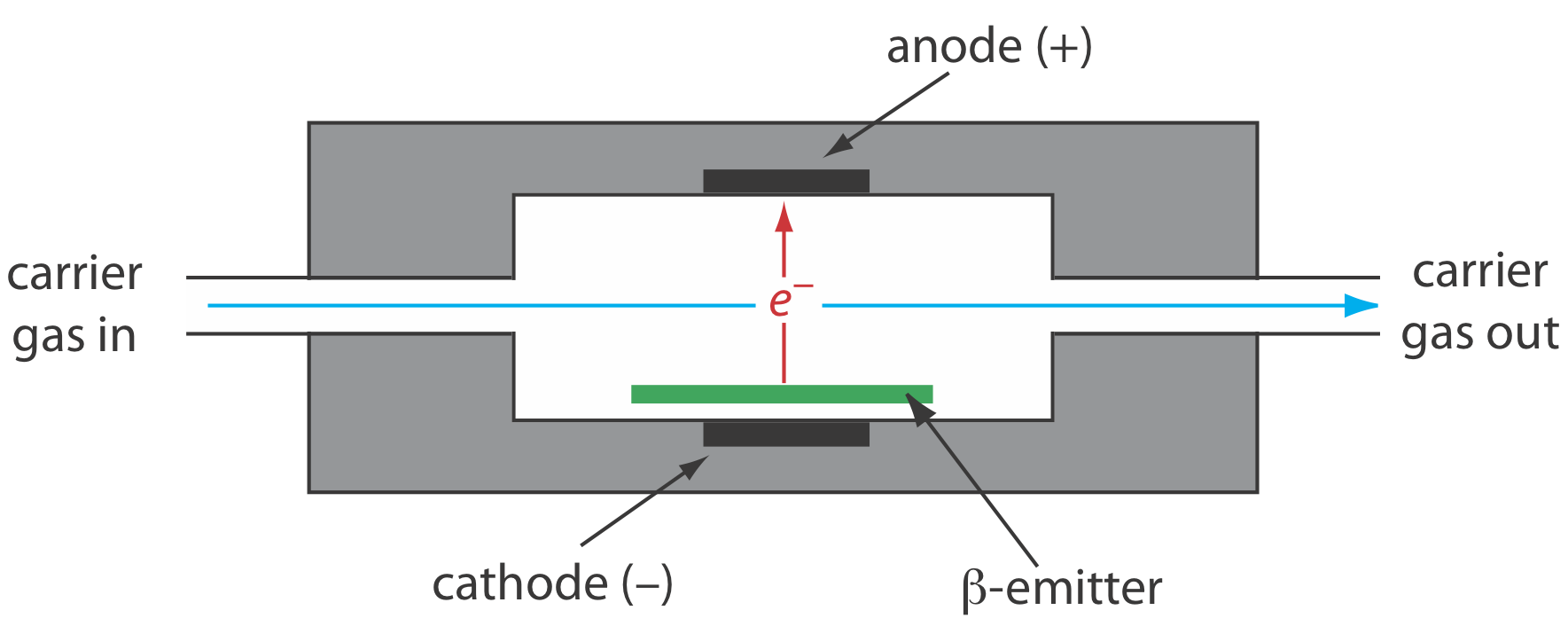
Детектор захоплення електронів є прикладом селективного детектора. Як показано на малюнку Template:index, детектор складається з\(\beta\) -випромінювача, такого як 63 Ni. Випромінювані електрони іонізують рухливу фазу, зазвичай N 2, генеруючи стоячий струм між парою електродів. Коли розчинене речовина з високою спорідненістю для захоплення електронів елюює з колони, струм зменшується, що служить сигналом. ECD є високоселективним до розчинених речовин з електронегативними функціональними групами, такими як галогени та нітрогрупи, і відносно нечутливий до амінів, спиртів та вуглеводнів. Хоча межа виявлення відмінна, його лінійний діапазон поширюється лише на два порядки.
А\(\beta\) -частинка - це електрон.
Мас-спектрометр (МС)
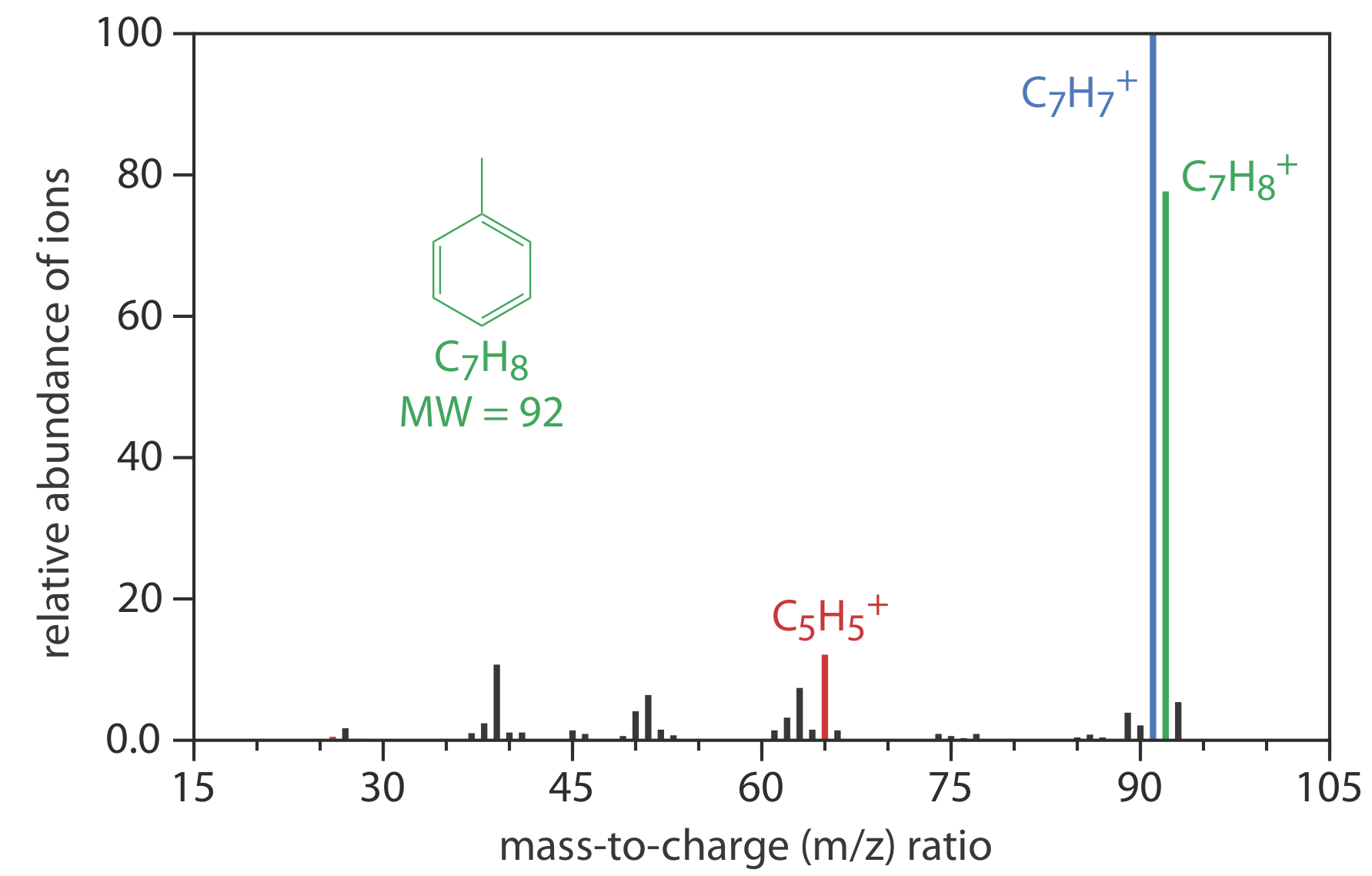
Мас-спектрометр - це прилад, який іонізує газоподібну молекулу, використовуючи достатню енергію, щоб отриманий іон розпадався на менші іони. Оскільки ці іони мають різне співвідношення маси до заряду, їх можна розділити за допомогою магнітного поля або електричного поля. Отриманий масовий спектр містить як кількісну, так і якісну інформацію про аналіті. На малюнку Template:index показано масовий спектр толуолу.
На малюнку Template:index показана блок-схема типового приладу газової хроматографії-мас-спектрометра (GC—MS). Стічні води з колони надходять у джерело іонів мас-спектрометра таким чином, що усуває більшу частину газу-носія. В іонізаційній камері залишилися молекули - суміш газу-носія, розчинника і розчинів - піддаються іонізації та фрагментації. Мас-аналізатор мас-спектрометра відокремлює іони за співвідношенням маси до заряду, а детектор підраховує іони та відображає масовий спектр.
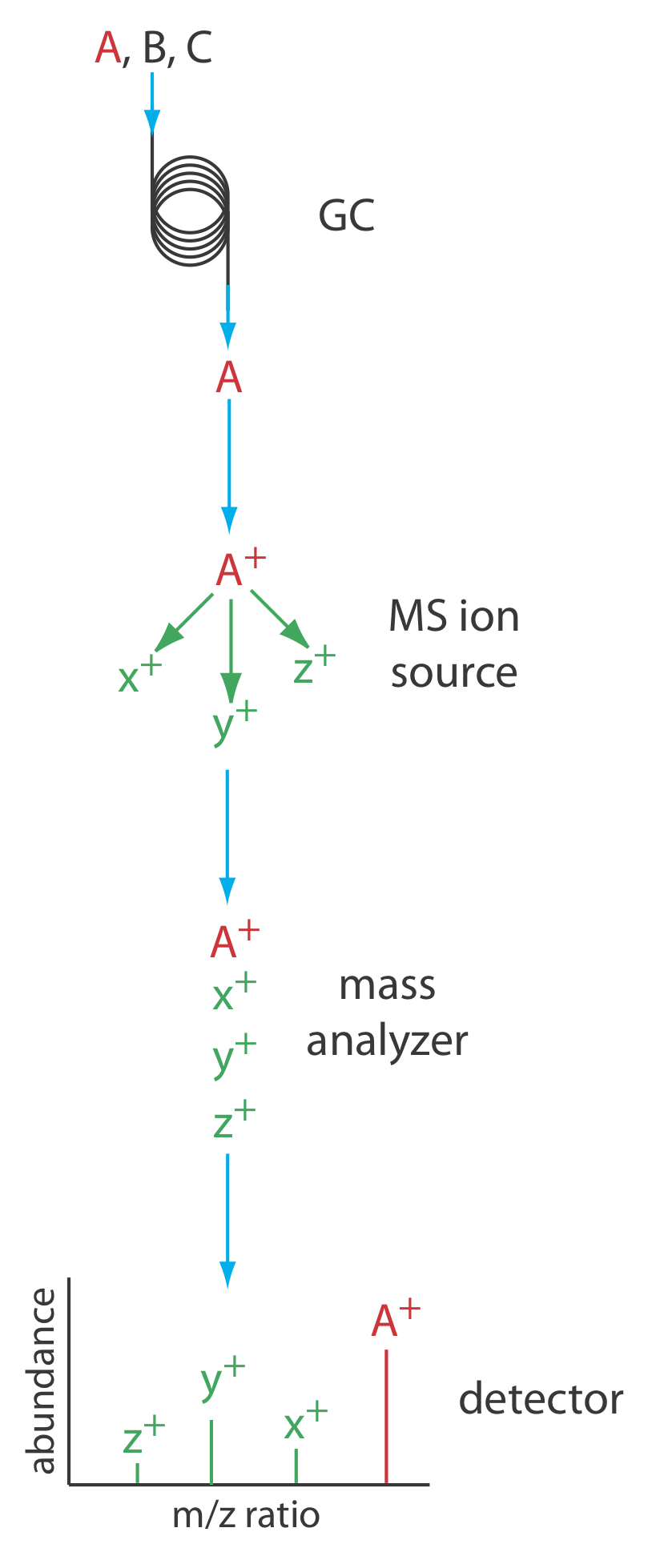
Існує кілька варіантів моніторингу хроматограми при використанні мас-спектрометра в якості детектора. Найпоширенішим методом є безперервне сканування всього масового спектру та повідомлення про загальний сигнал для всіх іонів, які досягають детектора під час кожного сканування. Це загальне іонне сканування забезпечує універсальне виявлення для всіх аналітів. Ми можемо досягти певної міри селективності, контролюючи одне або кілька конкретних коефіцієнтів маси до заряду, процес, який називається селективно-іонним моніторингом. Мас-спектрометр забезпечує відмінні межі виявлення, як правило, від 25 до 100 пг, з лінійним діапазоном 10 5 порядків. Оскільки ми постійно записуємо масовий спектр елюента колонки, ми можемо повернутися назад і вивчити масовий спектр для будь-якого приросту часу. Це є явною перевагою для GC—MS, оскільки ми можемо використовувати масовий спектр, щоб допомогти ідентифікувати компоненти суміші.
Інші детектори
Два додаткових детектора схожі за конструкцією на детектор іонізації полум'я. У фотометричному детекторі полум'я оптичне випромінювання фосфору та сірки забезпечує детектор селективним для сполук, що містять ці елементи. Термоіонний детектор реагує на сполуки, що містять азот або фосфор.
Інфрачервоний спектрофотометр з перетворенням Фур'є (FT-IR) також може служити детектором. У GC-FT-IR стоки з колони протікають через оптичну комірку, побудовану з трубки Pyrex розміром 10—40 см з внутрішнім діаметром 1-3 мм. Внутрішня поверхня клітини покрита відбиваючим шаром золота. Багаторазові відбиття випромінювання джерела, коли воно передається через комірку, збільшують довжину оптичного шляху через зразок. Як і у випадку з GC—MS, FT-ІЧ-детектор постійно записує спектр елюента колонки, що дозволяє нам досліджувати ІЧ-спектр для будь-якого приросту часу.